Гострий лімфобластний лейкоз (ГЛЛ)/лімфобластна лімфома (ЛБЛ)
Гострий лімфобластний лейкоз (ALL, ГЛЛ)/лімфобластна лімфома (ЛБЛ) – це захворювання клональних гемопоетичних стовбурових клітин В- або Т-клітинного походження. Система класифікації Всесвітньої організації охорони здоров’я (ВООЗ) 2017 року класифікує ці захворювання як попередники лімфоїдних новоутворень.
Лімфобласти є характерними клітинами цього захворювання. Діагноз ГЛЛ ставиться, коли кількість бластів перевищує 20%. Іноді у пацієнтів спостерігають первинне ураження лімфатичних вузлів або екстранодальних ділянок (LBL – Лімфобластна лімфома).
Епідеміологія
ГЛЛ – це в основному захворювання дітей молодше 6 років з невеликим переважанням чоловіків. Близько 85% випадків мають В-клітинне походження, а 15% – Т-клітинне походження.
На LBL припадає приблизно 2% усіх НХЛ. B-LBL становить приблизно 10% усіх випадків LBL, і переважна більшість (90%) є T-LBL. Т-клітинний ГЛЛ частіше зустрічається у чоловіків, афроамериканців і підлітків. T-ALL становить приблизно 25% дорослих ALL.
Орієнтовна щорічна захворюваність на гострий лімфобластний лейкоз (ГЛЛ) у всьому світі становить від 1 до 5 випадків/100 000 населення, і більше двох третин випадків ГЛЛ мають В-клітинний фенотип.
В-клітинна ГЛЛ/ЛБЛ – це переважно дитяче захворювання, причому три чверті випадків зустрічаються у дітей віком до 6 років; є другий пік захворюваності у дорослих старше 60 років.
В-клітинний ГЛЛ зустрічається трохи частіше у чоловіків, ніж у жінок. Захворюваність на B-клітинну ALL/LBL у три рази вища серед білих людей, ніж серед чорношкірих. Латиноамериканське населення має найвищий рівень захворюваності серед усіх етнічних груп, і захворюваність, як видається, зростає в деяких районах Центральної та Південної Америки (наприклад, Гватемала, Перу) з невідомих причин.
Причина B-клітинної ALL/LBL невідома, але вона може бути пов’язана з іонізуючою радіацією та/або ще невідомими інфекційними агентами. Справжній сімейний ALL зустрічається рідко, хоча він був пов’язаний із спадковими мутаціями PAX5, ETV6 і TP53. Існує підвищена частота В-клітинної ГЛЛ у дітей із синдромом Дауна та іншими конституційними розладами. В-клітинний ALL/LBL також асоціюється з певними однонуклеотидними поліморфізмами в конкретних генах, включаючи GATA3, ARID5B, IKZF1, CEBPE та CDKN2A/B.
Клінічні прояви
В-клітинний лімфобластний лейкоз і В-клітинна лімфобластна лімфома є збігаючими клінічними проявами одного захворювання.
Більшість пацієнтів мають ознаки, пов’язані з анемією, нейтропенією та/або тромбоцитопенією через ураження кісткового мозку. Кількість лейкоцитів може бути зниженою, нормальною або помітно підвищеною. Симптоми можуть включати втому, інфекції або легкі/спонтанні синці чи кровотечі. Біль у кістках або артралгії можуть бути явними скаргами, а конституційні симптоми (наприклад, лихоманка, нічна пітливість, ненавмисна втрата ваги) часто присутні, але, як правило, легкі. Гепатомегалія, спленомегалія та/або лімфаденопатія спостерігаються приблизно у половини дорослих. Ураження центральної нервової системи (ЦНС) може проявлятися краніальними невропатіями або менінгеальними симптомами.
Діагностика
B-клітинний ALL/LBL зазвичай підозрюють у дитини або дорослого з циркулюючими лімфобластами та/або безболісною лімфаденопатією, його можна запідозрити на фоні цитопенії нез’ясованої етіології; втоми, інфекції, легких або спонтанних синців/кровотеч; конституціональних симптомів; болю у кістках; та/або гепатомегалія або спленомегалія.
Оцінка повинна включати загальний аналіз крові з диференціальним дослідженням, дослідження периферичного мазка, імунофенотипування периферичної крові або кісткового мозку та дослідження кісткового мозку; у деяких випадках оцінка може включати ексцизійну або пункційну біопсію лімфатичного вузла.
Цей матеріал також слід проаналізувати цитогенетичними та молекулярними методами, щоб уможливити класифікацію відповідно до класифікації Всесвітньої організації охорони здоров’я (ВООЗ).
Діагностика B-клітинної ALL/LBL вимагає виявлення лімфобластів із характерним імунофенотипом у периферичній крові, кістковому мозку чи інших залучених тканинах; хоча немає консенсусу щодо мінімальної частки лімфобластів у кістковому мозку, слід уникати діагностики B-клітинної ALL/LBL, якщо лімфобластів <20% .
Лімфобластні новоутворення класифікуються на основі В-клітинного походження проти Т-клітинного походження:
- B-клітинний лімфобластний лейкоз/лімфома (B-клітинний ALL/LBL)
- Т-клітинний лімфобластний лейкоз/лімфома (Т-клітинний ALL/LBL)
Лейкемія та лімфома є накладанням клінічних проявів одного захворювання (тобто В-клітинний ALL/LBL); діагностика та класифікація не розрізняють ці захворювання.
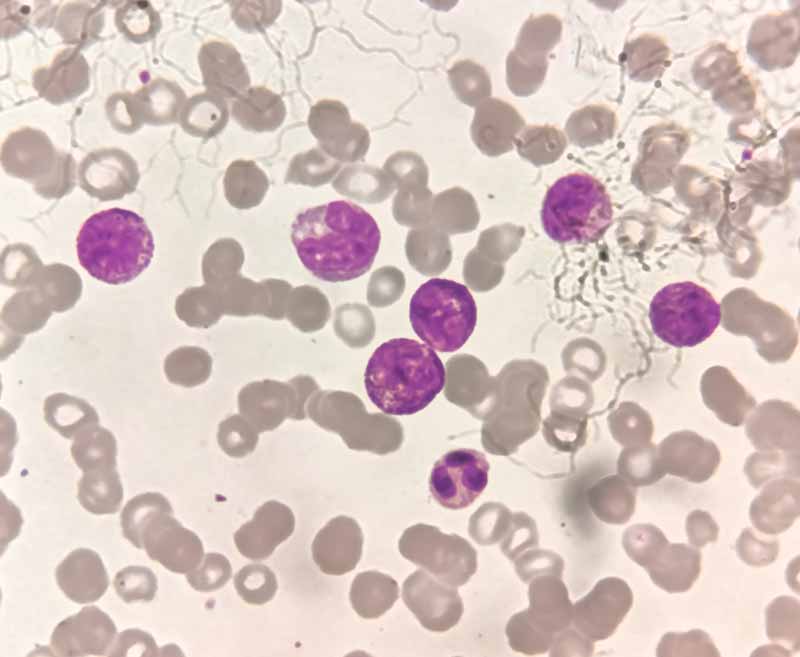
У мазку крові виявляються лімфобласти, які можуть варіюватися від дрібних клітин з невеликою цитоплазмою, конденсованим ядерним хроматином і нечіткими ядерцями до більших клітин з помірною кількістю цитоплазми, розсіяним хроматином і кількома ядерцями.
Можуть бути присутні кілька азурофільних цитоплазматичних гранул, але палички Ауера відсутні.
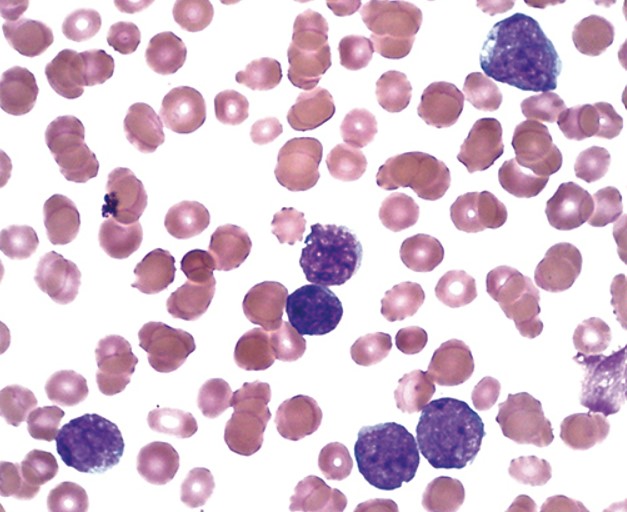
Лімфобласти в мазку периферичної крові
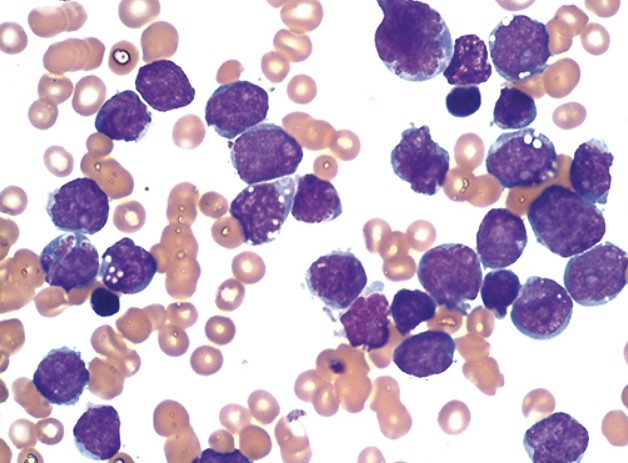
Лімфобласти в мазку кісткового мозку
Проточна цитометрія/імуногістохімія – імунофенотип, визначений за допомогою проточної цитометрії та/або імуногістохімії з циркулюючих лімфобластів, зразка кісткового мозку або матеріалу лімфатичних вузлів, необхідний для діагностики B-клітинної ALL/LBL.
Важливою характеристикою В-клітинної ALL/LBL є експресія В-клітинних антигенів за відсутності Т-клітинних антигенів; в деяких випадках може виявлятися мієлоїдний антиген:
- В-клітинні антигени – лімфобласти В-клітинного ALL/LBL майже завжди позитивні на В-клітинні маркери CD19, цитоплазматичний CD79a та цитоплазматичний CD22; жоден із цих маркерів окремо не є специфічним для діагнозу, але їх позитивність у комбінації або при високій інтенсивності переконливо підтверджує діагноз.
- У більшості випадків лімфобласти також позитивні на CD10, CD24, PAX5 і TdT (термінальна дезокситрансфераза); Експресія CD20 і CD34 варіабельна, а CD45 може бути відсутнім або тьмяним. Експресія CD20 присутня приблизно в 40-50 % всіх випадків В-клітин.
- Т-клітинні антигени – Т-клітинні антигени (наприклад, CD3) негативні.
- Мієлоїдні антигени – мієлоїдні антигени, такі як CD13 і CD33, можуть експресуватися, що не виключає діагноз В-клітинної ALL/LBL; однак експресія мієлопероксидази (MPO) вважається визначальною для мієлоїдної лінії та виключає діагноз.
Експресія CD13 та/або CD33 пов’язана з певними категоріями В-клітин ALL/LB (наприклад, ETV6::CBFA2 , KMT2A , BCR::ABL1 ).
Цитогенетичний/молекулярний аналіз – звичайне хромосомне смугування з або без флуоресцентної гібридизації in situ (FISH) і молекулярний аналіз (на перегрупування генів і моделі експресії генів) слід проводити на циркулюючих лімфобластах, зразках кісткового мозку та/або лімфатичних вузлах матеріал для класифікації B-клітинного ALL/LBL.
Флуоресцентна гібридизація in situ (FISH) для t(12;21) може бути важливою, оскільки цю перебудову важко розпізнати за допомогою каріотипування. З огляду на те, що наявність злитих генів KMT2A і BCR::ABL1 може визначати прогноз і рішення щодо лікування, для цих аномалій слід провести тест FISH.
Слід також провести тестування для виявлення BCR:ABL1 -подібного ALL, в ідеалі шляхом профілювання експресії генів. Альтернативні підходи (наприклад, скринінг масивів низької щільності [LDA], проточна цитометрія для виявлення надмірної експресії CRLF2 або виявлення злитих генів, асоційованих з BCR:ABL1 -подібним ALL за допомогою цитогенетики/FISH або секвенування РНК) також можна використовувати для виявлення багатьох випадків BCR :ABL1 -як ALL.
Перегрупування генів рецепторів антигенів у В-клітинних ALL/LBL є змінною і не є специфічною для лінії; як наслідок, виявлення таких перегрупувань не є надійним методом для визначення походження. Майже всі випадки В-клітинної ГЛЛ мають перебудову важкого ланцюга імуноглобуліну (IGH ), але до 70 % випадків також мають перебудову генів гамма- або бета-ланцюга рецептора Т-клітин (TCR).
Класифікація, різновиди
Усі випадки B-клітинної ALL/LBL слід класифікувати відповідно до імунофенотипічної та цитогенетичної/генетичної класифікації.
Немає необхідності розрізняти лейкемію та лімфому, оскільки вони вважаються різними клінічними проявами одного захворювання.
Класифікація пухлин кровотворної та лімфоїдної тканин Всесвітньої організації охорони здоров’я (ВООЗ) класифікує ALL/LBL відповідно до лімфоїдної лінії походження, цитогенетичних даних і молекулярних особливостей.
Класифікація ВООЗ 2016 року ALL/LBL базується на імунофенотипі, цитогенетичних аномаліях і молекулярних змінах.
ALL/LBL широко поділяють на пухлини В-клітин, Т-клітин і природних клітин-кілерів (NK) на основі імунофенотипу.
B-клітинний ALL/LBL, NOS – B-клітинний ALL/LBL, не специфікований іншим чином (NOS) не має жодної з ознак, які визначають різні цитогенетичні/генетичні категорії.
Ph+; t(9;22)(q34,1; q11,2); BCR::ABL1 – Філадельфійська (Ph) хромосома, t(9;22), пов’язана з експресією онкогену BCR::ABL1. Хоча Ph+ ALL історично вважався несприятливою ознакою, його прогностичне значення покращилося в еру інгібіторів тирозинкінази.
Ph виявляється приблизно в одній чверті дорослих ALL, але лише у 2-4 відсотках випадків у дітей; захворюваність зростає з віком і виявляється у 40-50 % пацієнтів ≥60 років.
Ph+ B-клітинний ALL/LBL визначається за допомогою хромосомних смуг, флуоресцентної гібридизації in situ (FISH) та/або виявлення BCR::ABL1 за допомогою RT-полімеразної ланцюгової реакції; приблизно половина випадків експресує білок p210 BCR::ABL1 (характерний для хронічного мієлоїдного лейкозу), а половина експресує білок p190.
t(v;11q23,3); KMT2A – rearranged – Ця категорія B-клітин ALL/LBL містить t(v;11q23.3) і транслокацію, що включає KMT2A (раніше називався MLL ) з одним із >100 партнерів злиття, включаючи AFF1 (AF4), MLLT1 (ENL) або MLLT3 (AF9).
Це найпоширеніша категорія B-клітинної ALL у немовлят; це рідше зустрічається у дітей старшого віку, але стає все більш поширеним у дорослому віці. Кількість лейкоцитів (лейкоцитів) зазвичай підвищується, часто >100 000/мкл. Ураження центральної нервової системи (ЦНС) є загальним і може спостерігатися інфільтрація інших органів, хоча чисті лімфоматозні прояви не є типовими.
KMT2A -перебудовані пухлини зазвичай демонструють про-В імунофенотип (CD19+, CD10-), часто з коекспресією мієлоїдних маркерів CD15, CD33 і CD68.
Лейкемії з делецією 11q23.3 без перегрупування KMT2A не входять до цієї категорії.
t(12;21)(p13.2;q22.1); ETV6::RUNX1 – ця категорія В-клітин ALL/LBL пов’язана з t(12;21) і злиттям ETV6::CBFA2 (раніше відомим як TEL::AML1).
Цей підтип є найпоширенішою формою B-клітинної ALL/LBL, пов’язаної з визначальною хромосомною перебудовою у дітей (приблизно одна чверть випадків); це не спостерігається у немовлят і рідко зустрічається в дорослому віці.
Ці пухлини зазвичай мають пре-В-клітинний імунофенотип (CD19+, CD10+). Відсутність або тьмяна експресія CD9, CD20 і CD66c є відносно специфічною для цього типу пухлин. Злоякісні клітини зазвичай є CD34+, і вони часто експресують мієлоїдні антигени (особливо CD13).
Ця форма B-клітинної ALL/LBL пов’язана з унікальною ознакою експресії генів. FISH може знадобитися для виявлення цього розладу, оскільки деякі перебудови є цитогенетично загадковими.
B-клітина ALL/LBL t(12;21)(p12;q22); ETV6::CBFA2 асоціюється з хорошим прогнозом.
Гіпердиплоїдія – гіпердиплоїдна В-клітина ALL/LBL має >50 хромосом, як правило, без транслокацій або інших структурних хромосомних змін. Гіпердиплоїдний B-клітинний ALL можна виявити за допомогою звичайного каріотипування, FISH або проточної цитометрії ДНК-індексу.
Гіпердиплоїдний В-клітинний ГЛЛ поширений у дітей (≥25 % випадків у дитинстві), він не спостерігається у немовлят, і його частота знижується серед дітей старшого віку. У дорослому віці (від 7 до 8 %) В-клітинна ГЛЛ зустрічається рідко.
Гіпердиплоїдний В-клітинний ALL/LBL не має морфологічних або клінічних ознак. Лімфобласти мають пре-В-клітинний імунофенотип (CD19+, CD10+) і в більшості випадків є CD34+; CD45 часто відсутній .
Прогностичне значення у дорослих невизначено, але гіпердиплоїдний В-клітинний ГЛЛ має дуже сприятливий прогноз у дітей.
Гіподиплоїдія – гіподиплоїдний B-клітинний ALL/LBL характеризується втратою однієї або кількох хромосом.
Близько 5 % випадків В-клітинної ALL/LBL є гіподиплоїдними, але лише 1 % має <45 хромосом. Захворюваність гіподиплоїдією вища у дорослих.
Лімфобласти гіподиплоїдних В-клітин ALL/LBL мають пре-В-клітинний імунофенотип (CD19+, CD10+), але немає інших відмінних імунофенотипічних або клінічних ознак.
Всі гіподиплоїдні В-клітинні випадки можуть бути розділені на основі кількості хромосом на категорії з характерними генетичними ураженнями: майже гаплоїдні (23-29 хромосом), низькі гіподиплоїдні (33-39), високі гіподиплоїдні (40-43) і майже- диплоїдний (від 44 до 46); Як приклад, майже гаплоїдний В-клітинний ALL часто має мутації RAS або рецепторної тирозинкінази, тоді як більшість низькогіподиплоїдного B-клітинного ALL мають мутації втрати функції TP53 та/або RB1.
Майже диплоїдні класи асоціюються з хорошим прогнозом, але всі інші класи гіподиплоїдних В-клітин ALL/LBL асоціюються з поганим прогнозом.
t(5;14)(q31.1;q32.1); IGH::IL3 – у цій категорії лімфобласти містять транслокацію між IL3 та IGH (важкий ланцюг імуноглобуліну).
На цей рідкісний розлад припадає <1 % випадків B-клітинної ГЛЛ, про нього повідомлялося як у дітей, так і у дорослих.
Форма B-клітинної ALL/LBL часто асоціюється з низькою кількістю циркулюючих лімфобластів, що супроводжується безсимптомною еозинофілією; еозинофіли є реактивною популяцією клітин, а не частиною злоякісного клону. Навіть невелика кількість CD19+, CD10+ бластів у пацієнта з еозинофілією переконливо свідчить про цей діагноз.
Перебудова між IL3 на хромосомі 5 та IGH на хромосомі 14 викликає конститутивну надекспресію IL3.
Вважається, що прогноз не відрізняється від інших категорій В-клітинної ГЛЛ.
t(1;19)(q23;p13,3); TCF3-PBX1 – наявність t(1;19) транслокації та/або злиття TCF3 (також відомого як E2A ) і PBX1 визначає цю категорію. Транслокація TCF3::PBX1 генерує злитий білок, який діє як активатор транскрипції, а також заважає нормальному функціонуванню TCF3 і PBX1.
Ця цитогенетична/генетична аномалія відносно поширена у дітей (приблизно 6 % B-клітинної ОЛЛ), але рідше зустрічається у дорослих.
Лімфобласти зазвичай мають пре-В фенотип і є CD19+, CD10+, CD9+, експресують цитоплазматичний важкий ланцюг Mu та є тьмяними або негативними для CD34. Ця категорія не має унікальних морфологічних або клінічних аспектів, але має чіткий профіль експресії генів. У цих пацієнтів може бути підвищений ризик рецидиву ЦНС.
Категорія В-клітин ALL/LBL t(1;19)(q23;p13.3); TCF3::PBX1 не має особливого прогностичного значення при сучасному лікуванні.
Демонстрація лише перебудови TCF недостатня для діагностики цієї категорії лейкемії, тому що альтернативна транслокація TCF3, яка включає ген HLF , пов’язана з t(17;19) і пов’язана з несприятливим прогнозом.
BCR::ABL1 -подібний (Ph-подібний) – ця категорія пов’язана з сигнатурою експресії гена, яка нагадує сигнатуру ALL Ph+ B-клітин, але в клітинах відсутні характерна (9;22) транслокація та пов’язаний злитийген BCR::ABL1.
На BCR ::ABL1 -подібну категорію припадає приблизно 10 % дитячих B-клітинних ALL/LBL, але до однієї третини випадків у дорослих.
Пацієнти з BCR::ABL1 -подібним B-клітинним ALL/LBL, як правило, мають високу кількість лейкоцитів при зверненні. Лімфобласти мають фенотип CD19+, CD10+.
Випадки з транслокаціями CRLF2 показують високий рівень експресії поверхневого CRLF2 за допомогою проточної цитометрії, але немає специфічних імунофенотипових особливостей, пов’язаних з іншими молекулярними аномаліями. Деякі перебудови можна виявити за допомогою традиційного аналізу каріотипу, але інші є загадковими. Діагностика цього стану зазвичай вимагає аналізу експресії генів у поєднанні з цитогенетичним і молекулярним діагностичним тестуванням.
Приблизно в половині цих випадків є транслокації за участю CRLF2, тоді як інші мають транслокації за участю тирозинкіназ (наприклад, JAK2 , ABL1, ABL2, PDGFRB, NTRK3, TYK2, CSF1R) або активації EPOR. Значна частка цих пухлин також має мутації в IKZF1, гені, який кодує важливий транскрипційний регулятор розвитку лімфоїдів. Частота специфічних перебудов залежить від віку. Перегрупування CRLF2 є найпоширенішими змінами в усіх вікових групах. Злиття ABL частіше зустрічаються у дітей, а перегрупування JAK2 частіше зустрічаються у молодих людей
Ця категорія B-клітинної ALL/LBL асоціюється з поганим прогнозом, а випадки з транслокаціями CRLF2 мають особливо поганий прогноз.
iAMP21 – Ампліфікація частини хромосоми 21 зазвичай виявляється зондом FISH для RUNX1, який виявляє ≥5 копій гена (або ≥3 додаткових копій на одній аномальній хромосомі 21). Ця аномалія менш поширена у дорослих. В-клітинний ALL/LBL підвищується в 3000 разів у осіб із конституційною Робертсонівською транслокацією rob(15;21)(q10;q10) через хромотрипсис; цей механізм також може сприяти спорадичним випадкам iAMP21.
На частку цього утворення припадає приблизно 2 % B-клітинної ГЛЛ і зазвичай виявляється у дітей старшого віку, особливо у тих, у кого низька кількість лейкоцитів.
У більшості випадків iAMP21 пов’язаний з іншими хромосомними змінами. Це пов’язано з поганим прогнозом у дітей, хоча більш інтенсивна терапія може подолати цей несприятливий ризик.
Інші генетичні підтипи – Молекулярна та генетична оцінка когорти з майже 2000 випадків дитячого та дорослого B-клітинного ALL/LBL виявила додаткові чіткі генетичні підтипи, які ще належить включити до класифікації ВООЗ.
До них належать підтипи, визначені структурними перебудовами в генах DUX4, NUTM1, MEF2D, ZNF384 і MYC, а також інші генетичні аберації, пов’язані з PAX5 і IZKF1. Найбільш поширений з цих підтипів, визначений різноманітний PAX5 зміни, становили 7 % випадків у цій серії та мали ознаки, пов’язані із захворюванням високого ризику у дітей. Потрібні додаткові дослідження для оцінки цих нових генетичних підтипів, які, ймовірно, будуть включені в майбутні класифікації ВООЗ.
Класифікація ВООЗ гострого лімфобластного лейкозу
ВООЗ класифікує ALL як В-лімфобластний лейкоз/лімфому або Т-лімфобластний лейкоз/лімфому.
B лімфобластний лейкоз/лімфома:
- B-лімфобластний лейкоз/лімфома, не уточнений
- В-лімфобластний лейкоз/лімфома з рецидивуючими генетичними аномаліями
- B-лімфобластний лейкоз/лімфома з t(9;22)(q34.1;q11.2); BCR-ABL1
- B-лімфобластний лейкоз/лімфома з t(v;11q23.3); KMT2A
- B-лімфобластний лейкоз/лімфома з t(12;21)(p13.2;q22.1); ETV6-RUNX1
- В-лімфобластний лейкоз/лімфома з гіпердиплоїдією
- В-лімфобластний лейкоз/лімфома з гіподиплоїдією
- B-лімфобластний лейкоз/лімфома з t(5;14)(q31.1;q32.3) IL3-IGH
- B-лімфобластний лейкоз/лімфома з t(1;19)(q23;p13.3); TCF3-PBX1
- Попередньо: B-лімфобластний лейкоз/лімфома, BCR-ABL1- подібний
- Попередньо: B-лімфобластний лейкоз/лімфома з iAMP21
Т-лімфобластний лейкоз/лімфома:
- Попередньо: ранній Т-клітинний попередник лімфобластного лейкозу
- Попередньо: природний кілер (NK) лімфобластний лейкоз/лімфома
FAB-класифікація гострого лімфобластного лейкозу
- ALL-L1: дрібні клітини з гомогенним ядерним хроматином, правильною формою ядра, малими або відсутніми ядерцями, мізерною цитоплазмою та від легкої до помірної базофілії.
- ALL-L2: великі гетерогенні клітини зі змінним ядерним хроматином, неправильною формою ядра, 1 або більше ядерцями, різною кількістю цитоплазми та різною базофілією
- ALL-L3: Великі однорідні клітини з тонким хроматином із крапками; регулярні ядра; видатні ядерця; і рясна, глибоко базофільна цитоплазма.
Диференційна діагностика
При проведенні диференційної діагностики слід враховувати:
Бластна криза ХМЛ
Дуже важливо відрізнити PH-позитивний ГЛЛ від хронічного мієлоїдного лейкозу (ХМЛ) при бластному кризі.
ГЛЛ, що трансформується з мієлодиспластичного синдрому, все ще вважається ГЛЛ, але у тих, хто трансформується з інших мієлопроліферативних або мієлодиспластичних/мієлопроліферативних новоутворень, корисно знати, що гостре захворювання виникло внаслідок основного хронічного захворювання.
Лімфома Беркітта – лімфома Беркітта (BL) – це високоагресивна В-клітинна неходжкінська лімфома, яка може проявлятися у вигляді пухлини, що швидко зростає (наприклад, пухлина щелепи чи черевної порожнини) та/або лейкемія. BL може мати істотнe клінічну та морфологічну схожість з ALL/LBL. Гістологія БЛ зазвичай виявляє високопроліферативні мономорфні клітини середнього розміру з базофільною цитоплазмою (часто має вигляд «зоряного неба») , які, як правило, більші за лімфобласти ALL/LBL. Діагноз BL базується на морфології, імунофенотипі та цитогенетичних/молекулярних особливостях і відрізняється від ALL/LBL характерним імунофенотипом В-клітин зрілого зародкового центру та транслокацією, що включає хромосому 8 та/або MYC перегрупування, як обговорюється окремо.
Інші гострі лейкемії – лімфобласти ALL/LBL важко відрізнити морфологічно від інших форм гострого лейкозу, особливо тих, які є мінімально диференційованими:
- Гострий мієлоїдний лейкоз (ГМЛ) – мієлобласти ГМЛ зазвичай є незрілими клітинами з великими ядрами, помітними ядерцями та різною кількістю блідо-блакитної цитоплазми (іноді зі слабкою грануляцією та/або паличками Ауера). Клітини AML зазвичай фарбуються на мієлопероксидазу (MPO) або лізоцим і зазвичай є негативними на B- і T-клітинні антигени та TdT; деякі випадки AML експресують антигени, пов’язані зі стовбуровими клітинами або лімфоїдними лініями.
- Гостра недиференційована лейкемія (AUL) – бластні клітини AUL є морфологічно м’якими і загалом не відрізняються від клітин B-ALL/LBL. Ці утворення розрізняють за імунофенотипом; Бласти AUL не експресують специфічні для лінії антигени за допомогою проточної цитометрії чи імуногістохімії, тоді як бласти ALL/LBL експресують B- або T-лімфоїдні антигени.
- Гостра лейкемія зі змішаним фенотипом (MPAL) – Діагностика MPAL вимагає демонстрації як мієлоїдних, так і лімфоїдних маркерів за допомогою проточної цитометрії та/або імуногістохімічного дослідження.
Хронічний мієлоїдний лейкоз (ХМЛ) – ХМЛ зазвичай проявляється як розширена популяція мієлоїдних клітин на різних стадіях диференціювання з характерним t(9;22) (філадельфійська хромосома) з перегрупуванням BCR – ABL1, що може супроводжуватися спленомегалією та/або конституціональними симптомами.
Проте приблизно 10 % ХМЛ у бластній фазі можуть мати домінуючу популяцію лімфобластів. ХМЛ можна відрізнити від ALL/LBL шляхом виявлення t(9;22) або BCR-ABL1 у мієлоїдних клітинах.
- Мієлоїдні/лімфоїдні новоутворення, пов’язані з еозинофілією та перегрупуванням PDGFRA, PDGFRB або FGFR1 або PCM1-JAK2 – ці рідкісні новоутворення часто супроводжуються лімфаденопатією внаслідок проліферації лімфобластів Т-лінії (або рідше В-лінії), які морфологічно та імунофенотипово не відрізняється від звичайного LBL. Характерні перебудови генів також присутні в мієлоїдних клітинах, а також у лімфобластах, і ці порушення слід запідозрити, якщо дослідження крові виявляє лейкоцитоз та еозинофілію. Вони діагностуються на основі ідентифікації одного з патогенних перебудов генів (перелічених вище), як правило, за допомогою флуоресцентної гібридизації in situ (FISH).
- Апластична анемія – Апластична анемія (АА) може проявлятися у дітей у вигляді панцитопенії з ретикулоцитопенією (часто <10 000/мкл) і глибоким гіпоцелюлярним ураженням кісткового мозку зі зниженням усіх кровотворних елементів. На відміну від ALL/LBL, при АА відсутній інфільтрат злоякісних клітин; замість цього простір кісткового мозку в основному складається з жирових клітин і строми кісткового мозку.
- Невеликі круглі синьоклітинні пухлини. У дітей невеликі круглі синьоклітинні пухлини, включаючи саркому Юінга (ES) і периферичну примітивну нейроектодермальну пухлину (PNET), можуть морфологічно нагадувати B-ALL/LBL, але ці захворювання відрізняються рівномірною популяцією невеликих, круглих, блакитних клітин з гіперхроматичними ядрами та мізерною цитоплазмою; відсутність В-лімфоїдних маркерів; може включати певні цитогенетичні/молекулярні зміни (наприклад, t(11;22)(q24;q12), перегрупування EWSR1 – FLI1 ).
- Незлоякісні розлади – Ознаки та симптоми ALL/LBL є неспецифічними, і певні незлоякісні захворювання можуть клінічно нагадувати ALL/LBL та/або виявляти лімфоцити, які можуть нагадувати лімфобласти.
У диференційну діагностику можуть бути включені такі захворювання:
Імунна тромбоцитопенія (ІТП) пов’язана з ізольованою тромбоцитопенією, яка клінічно може нагадувати ALL/LBL.
Діагноз ІТП у здорової на вигляд людини з типовим проявом (тобто раптова поява петехіального висипу або синців із ізольованою тромбоцитопенією) є простим. Тромбоцитопенія при ІТП, особливо в контексті нормального загального аналізу крові, як правило, є більш серйозною, ніж при ALL/LBL.
Навпаки, тромбоцитопенія при ALL/LBL майже завжди пов’язана з іншими клінічними та лабораторними результатами (наприклад, лихоманка, біль у кістках, втрата ваги, гепатоспленомегалія, лімфаденопатія, лейкоцитоз, анемія, лімфобласти периферичної крові).
Інфекційне захворювання та інші причини лімфоцитозу та/або аномальних лімфоцитів включають:
- Вірус імунодефіциту людини (ВІЛ)
- Інфекційний мононуклеоз
- Коклюш
- Остеомієліт
- Туберкульоз
- Токсичність важких металів
- Тимома
- Аутоімунні захворювання (включаючи ювенільний ревматоїдний артрит у дітей)
Прогноз
Прогноз при B-клітинній ALL/LBL пов’язаний з віком, клінічними особливостями та цитогенетичними/генетичними особливостями.
Клінічні ознаки, які пов’язані з несприятливим прогнозом, включають:
- Вік (≤1 рік або ≥10 років)
- Багато немовлят мають транслокації за участю KMT2A ( MLL ) на 11q23, що асоціюється з поганим прогнозом у будь-якому віці.
- Діти старшого віку частіше мають гіпердиплоїдію та t(12;21), що забезпечує кращий прогноз.
- ALL/LBL у дорослих В-клітин частіше асоціюється з аномаліями t(9;22) або низьким ризиком t(v;11q23.3), а виживаність нижча, ніж у дитячому віці.
- Кількість лейкоцитів ≥50 000/мкл
- Раса (іспаномовна або темношкіра) через геномні варіації та соціально-економічні фактори
- Чоловіча стать
Лікування
Гострий лімфобластний лейкоз (ГЛЛ) із позитивною філадельфійською хромосомою (Ph+) є біологічно та клінічно відмінною формою, класифікованою як ГЛЛ з t(9;22)(q34;q11.2); BCR-ABL1 у системі класифікації Всесвітньої організації охорони здоров’я (ВООЗ).
Експресія тирозинкінази BCR-ABL1 відрізняє Ph+ ALL від інших типів ALL і робить цю лейкемію чутливою до лікування інгібітором тирозинкінази BCR-ABL1 (TKI), який є важливим компонентом лікування. Терапія індукції ремісії призначена для досягнення повної гематологічної ремісії та надійної молекулярної відповіді. Ця фаза лікування повинна супроводжуватися підтримуючою терапією після ремісії, щоб забезпечити довготривалий контроль захворювання та можливе лікування.
Лікування вперше діагностованого гострого лімфобластного лейкозу (ГЛЛ) із позитивною філадельфійською хромосомою (Ph+) у дорослих.
| Індукційна терапія |
| Інгібітор тирозинкінази BCR-ABL (TKI) плюс одна з наступних комбінованих схем хіміотерапії: – Рак і лейкемія Група B (дослідження CALGB 8811 або 9111) Схема ALL – Гіперфракціонований циклофосфамід, вінкристин, доксорубіцин і дексаметазон (Hyper-CVAD) чергування з високими дозами метотрексату та цитарабіну – Французька схема GRAALL-2003 – Німецькі протоколи GMALL 06/99 та 7/03 Комбінація ІТК з кортикостероїдом або без нього здатна призвести до повної ремісії при Ph+ ALL, які раніше не лікувалися, вона має низьку токсичність. Ця комбінація не порівнювалася безпосередньо з ІТК плюс хіміотерапія. Якщо початкові дані підтвердяться подальшими дослідженнями, комбінація ІТК і стероїдів може стати відмінним варіантом для пацієнтів, наприклад літніх людей, які не можуть переносити комбіновану хіміотерапію. |
| Консолідація |
| Перевага віддається алогенній трансплантації гемопоетичних клітин у першій повній ремісії. Якщо пацієнт не є кандидатом на трансплантацію, слід провести консолідаційну хіміотерапію, яка включає BCR-ABL TKI. |
| Підтримуюча терапія |
| BCR-ABL TKI протягом двох років після алогенної трансплантації гемопоетичних клітин або на невизначений термін, якщо трансплантація не виконана. |
| Спостереження |
| Оцінка кісткового мозку та тестування на мінімальну залишкову хворобу з транскриптами BCR-ABL за допомогою кількісної полімеразної ланцюгової реакції в реальному часі. Примітка: обраний BCR-ABL TKI має відповідати тирозинкіназі BCR-ABL, яка використовується в конкретному протоколі лікування. |
Перед лікуванням пацієнта слід обстежити на наявність супутніх захворювань, неврологічних симптомів і критичної цитопенії.
Необхідно підтвердити наявність BCR-ABL1, оцінити придатність за станом здоров’я та встановити центральний венозний катетер. Перед початком індукційної терапії слід стабілізувати медичні ускладнення (наприклад, інфекції, кровотечі).
Клінічна/лабораторна оцінка
- Клінічні:
- Ускладнення та наслідки лейкемії. Пацієнта слід обстежити на наявність ускладнень та наслідків анемії (наприклад, задишки, слабкості), нейтропенії (наприклад, інфекції) та тромбоцитопенії (наприклад, кровотечі, синці).
- Супутні захворювання – слід оцінити клінічні прояви супутніх захворювань, включаючи захворювання серця, легеневі процеси та ниркову недостатність, оскільки вони можуть вплинути на вибір індукційної терапії.
- Неврологічна оцінка. Пацієнти з головними болями, змінами зору, слабкістю або іншими неврологічними симптомами або ознаками (наприклад, краніальні нейропатії, моторні або сенсорні зміни, атаксія) повинні пройти візуалізацію та люмбальну пункцію (ЛП), щоб визначити, чи ураження центральної нервової системи (ЦНС) пов’язані з ураженням лейкемією. Зауважте, що якщо ЛП не виконується як частина оцінки перед лікуванням, терапевтична ЛП для проведення профілактичної інтратекальної хіміотерапії має збігатися з початком індукційної терапії ремісії, як описано нижче.
- Лейкостаз. Пацієнти з легеневими, неврологічними або серцевими патологіями на тлі гіперлейкоцитозу (наприклад, кількість лейкоцитів >200 000/мікрол) повинні бути оцінені на лейкостаз і лікуватися, як обговорюється окремо.
- Лабораторна оцінка:
- Загальний аналіз крові з диференціалом, протромбіновий час (ПЧ), частковий тромбопластиновий час (ПЧТ), фібриноген, D-димер.
- Хімічні аналізи сироватки, включаючи електроліти, глюкозу, функції нирок і печінки, лактатдегідрогеназу (ЛДГ), кальцій, фосфор, сечову кислоту, альбумін і загальний білок.
- Серологічне дослідження гепатиту В і С, вірусу простого герпесу (ВПГ) і цитомегаловірусної (ЦМВ) інфекції.
- Типування людського лейкоцитарного антигену (HLA) слід проводити для пацієнтів, які є кандидатами на майбутню трансплантацію гемопоетичних клітин (HCT).
- Тест на вагітність для жінок дітородного віку.
- Візуалізація:
- Серцева функція – вихідне дослідження фракції викиду за допомогою ехокардіограми або радіонуклідної вентрикулограми. Ведення пацієнтів із зниженою серцевою функцією (наприклад, фракція викиду <40 %, значні аритмії) обговорюється нижче.
- Неврологічна візуалізація. Як зазначалося вище, пацієнти з неврологічними ознаками повинні пройти візуалізацію та ЛП, щоб визначити, чи пов’язані вони з лейкемічним ураженням ЦНС.
- Інші візуалізації слід виконувати, як це виправдано, для дослідження інфекцій чи інших наслідків лейкемії чи супутніх захворювань.
Перед початком терапії слід провести цитогенетичне та молекулярне тестування, щоб відрізнити Ph+ ALL від інших станів і підтвердити виявлення BCR-ABL1, щоб встановити базову лінію для моніторингу вимірюваної залишкової хвороби (MRD).
- Дослідження кісткового мозку, включаючи гістологічну оцінку досвідченим гематопатологом, цитогенетику, імунофенотип і молекулярний аналіз на BCR-ABL1 слід провести під час встановлення діагнозу, щоб виключити ALL з негативним філадельфійським хромосомою, хронічний мієлоїдний лейкоз (ХМЛ) у фазі лімфоїдного бластного кризу, та інші стани в диференціальній діагностиці.
- Необхідно провести кількісну ПЛР у реальному часі (RQ-PCR) для транскрипту BCR-ABL1, щоб встановити базове значення для моніторингу MRD.
- Аналіз мутацій BCR-ABL1 зазвичай не проводиться на початку індукційної терапії.
Медична придатність – перед початком терапії ми оцінюємо медичну придатність на основі стану працездатності, супутніх захворювань і фізіологічної придатності. Важливо, що вік сам по собі не визначає рівень фізичної підготовки.
Розглядаючи супутні захворювання, хронічним захворюванням слід приділяти більшу вагу, ніж тимчасовим станам, спричиненим ускладненнями лейкемії (наприклад, інфекцією, серцевою недостатністю, що посилюється анемією), які можуть покращитися за допомогою ефективної індукційної терапії та можуть підвищити толерантність до наступної терапії.
Інструменти для оцінки придатності за станом здоров’я:
- Функціональний статус Східної кооперативної онкологічної групи (ECOG).
- Фізіологічна придатність (наприклад, супутні захворювання, повсякденна діяльність, тести фізичної працездатності, когнітивні здібності), виміряна індексом супутніх захворювань Чарлсона (CCI).
- Класифікація придатності за станом здоров’я
- Медична придатність: як ECOG PS 0 до 2 , так і CCI 0 до 2
- Медично непридатний, але не слабкий: ECOG PS ≥3 або CCI ≥3
- Слабкий: І ECOG: ≥3 , і CCI ≥3
Терміни лікування – важливо контролювати ускладнення лейкемії та стабілізувати медичний стан пацієнта перед початком індукційної терапії ремісії. Може знадобитися кілька днів для отримання результатів цитогенетичних та/або молекулярних досліджень для підтвердження діагнозу Ph+ ALL та виключення інших станів у диференціальній діагностиці.
Очікуючи патологічного підтвердження діагнозу Ph+ ALL, можна лікувати інфекції, кровотечі, гіперурикемію, дегідратацію, ниркову дисфункцію, анемію, тромбоцитопенію та інші ускладнення.
Зазвичай може бути рекомендована гідратацію та алопуринол, щоб зменшити ризик гіперурикемії та її ускладнень. Якщо спостерігається виражений лейкоцитоз (наприклад, >50 000 лейкоцитів/мікрол), може бути рекомендоване поступове збільшення глюкокортикоїду (наприклад, починаючи з преднізону 25 мг на день) перед початком індукційної терапії. Деякі центри регулярно вводять глюкокортикоїди протягом трьох днів і проводять моніторинг синдрому лізису пухлини в очікуванні підтвердження діагнозу Ph+ ALL, але роль такого попереднього лікування ALL у дорослих не була чітко встановлена.
Необхідно вставити пристрій центрального венозного доступу.
Консультації
Направлення до групи трансплантологів для оцінки придатності для HCT та початку пошуку сімейного або альтернативного донора.
Консультування з питань фертильності та збереження.
Індукція ремісії
Пацієнта з Ph+ ALL слід або направити до спеціалізованого центру для надання медичної допомоги, або його повинен лікувати клініцист, який має необхідний досвід і ресурси для лікування з суворим дотриманням сучасного опублікованого протоколу. Після вибору схеми лікування важливо дотримуватися опублікованого протоколу, а не вибирати компоненти з різних протоколів лікування.
Основні компоненти – Існує консенсус, що всі схеми індукції ремісії для Ph+ ALL повинні включати:
●BCR-ABL1 інгібітор тирозинкінази (TKI).
плюс
●Глюкокортикоїди або хіміотерапія.
Інгібітор тирозинкінази
Для терапії індукції ремісії Ph+ ALL рекомендовано схему, яка включає ІТК, а не схему, яка не включає ІТК, ґрунтуючись на кращих показниках відповіді та віддалених результатах із невеликою додатковою токсичністю, коли ІТК включені.
Жодне рандомізоване дослідження не порівнювало конкретні режими індукції ремісії з або без ІТК. Проте включення ІТК в індукційну терапію пов’язане з помітно кращими результатами та незначною додатковою токсичністю, що базується на численних проспективних і ретроспективних дослідженнях.
Важливо, що ІТК є важливим компонентом індукційної терапії Ph+ ALL, але одного цього недостатньо. Оскільки ІТК сам по собі не забезпечує достатньо глибокої та/або стійкої молекулярної відповіді, його необхідно комбінувати з хіміотерапією або глюкокортикоїдом.
Вибір ІТК – немає специфічних ІТК BCR-ABL1, які є оптимальними для всіх пацієнтів із Ph+ ALL. Вибір ІТК залежить від профілю токсичності, супутніх захворювань, простоти введення, доступності та вартості. ІТК не порівнювалися прямо в Ph+ ALL, а різні режими індукції не були спеціально перевірені для кожного ІТК.
Дазатиніб має відомий досвід у проспективних дослідженнях і може певним чином проникати в ЦНС, він ефективний проти деяких мутацій BCR-ABL1, які не реагують на іматиніб, і, як правило, добре переноситься.
Лікування іншим ІТК може проводитися з таких причин:
- Наявні протипоказання до дазатинібу.
- Дазатиніб не схвалений до використання в певному регіоні.
Пацієнтам із плевральним випотом, легеневою гіпертензією, серцевими аритміями або якісними порушеннями тромбоцитів зазвичай призначають іматиніб, нілотиніб або понатініб.
Управління з харчових продуктів і медикаментів США (FDA) схвалило дазатиніб для Ph+ ALL, але він схвалений не в усіх країнах. Іматиніб, схвалений Європейським агентством з лікарських засобів (EMA) для початкового лікування Ph+ ALL, є прийнятною альтернативою.
Окремі ІТК не порівнювалися прямо в Ph+ ALL. Мета-аналіз та аналіз балів зі збігом зі схильністю показали перевагу схем на основі понатінібу над схемами, що містять ІТК першого або другого покоління, але понатініб пов’язаний із значною серцево-судинною токсичністю.
Лікування ІТК слід розпочинати під час встановлення діагнозу та продовжувати шляхом індукції ремісії та лікування після ремісії, якщо немає непереносимості або доказів стійкості до захворювання. Безперервна дія ІТК пов’язана з кращими результатами порівняно з імпульсним або періодичним лікуванням.
Побічні ефекти необхідно ефективно контролювати, щоб досягти оптимальної безпеки пацієнта, комфорту та результатів. Усі ІТК пов’язані з певними ранніми несприятливими ефектами, які можуть включати висип, нудоту, м’язові судоми, набряки, діарею або втому. Більшість із цих ранніх несприятливих ефектів є помірними, самообмеженими, і ними можна керувати за допомогою підтримуючих заходів.
Дазатиніб
Дазатиніб – є багатоцільовим ІТК, який широко використовується для Ph+ ALL. Лікування пов’язане з цитопеніями, плевральним випотом/затримкою рідини, подовженням інтервалу QTc та кровотечею. Слід уникати застосування дазатинібу пацієнтам із кровотечею в анамнезі, плевральним випотом або серцевою недостатністю.
Дазатиніб призначають у дозі 140 мг перорально щодня незалежно від прийому їжі; таблетки не слід подрібнювати або розрізати. Пацієнти повинні пройти скринінг за допомогою електрокардіограми (ЕКГ) для визначення інтервалу QTc на початковому рівні, а гіпокаліємія або гіпомагніємія повинна бути скоригована перед застосуванням дазатинібу. Пацієнтам з порушенням функції печінки або нирок корекція початкової дози не потрібна. Рекомендується дотримуватися обережності пацієнтам, які приймають інші лікарські засоби, які можуть призвести до подовження інтервалу QTc, а також сильні індуктори або інгібітори CYP3A4.
Більшість побічних ефектів дазатинібу є легкими та проходять самостійно. Проте у пацієнтів може спостерігатися подовження інтервалу QTc, затримка рідини (побічні ефекти ≥3 ступеня приблизно у 4 %), загострення застійної серцевої недостатності або значна кровотеча. Повідомлялося про серйозні та потенційно смертельні крововиливи з боку ЦНС та шлунково-кишкового тракту приблизно у 1 та 4 % пацієнтів відповідно; більшість випадків кровотечі були пов’язані з тяжкою тромбоцитопенією, антикоагулянтами та/або інгібіторами функції тромбоцитів.
У поєднанні з хіміотерапією дазатиніб зазвичай пов’язаний із >90% CR (Complete Response – Повна відповідь). Наприклад, індукція ремісії за допомогою дазатинібу та глюкокортикоїду у 63 пацієнтів – 98 % досягли CR та 29 % молекулярної повної ремісії. Також повідомлялося про високі показники CR і стійку молекулярну відповідь для дазатинібу в поєднанні з хіміотерапією низької інтенсивності, хіміотерапією високої інтенсивності і різними схемами хіміотерапії.
Дазатиніб активний проти багатьох мутацій домену кінази BCR-ABL1, які можуть бути присутніми на низьких рівнях під час діагностики, але він не активний проти мутації T315I (проти якої, як правило, ефективний лише понатиніб).
ІТК плюс хіміотерапія.
Вибір режиму хіміотерапії – для режиму індукції ремісії Ph+ ALL, що включає хіміотерапію, рекомендовано режим хіміотерапії низької або середньої інтенсивності, а не режим хіміотерапії високої інтенсивності. Це припущення ґрунтується на кращих результатах, меншій токсичності та меншій кількості смертей, пов’язаних із лікуванням, у дослідженні, у якому пацієнтів випадковим чином розподіляли на хіміотерапію низької інтенсивності проти хіміотерапії високої інтенсивності.
Дослідження фази 3 випадковим чином розподіляло 268 дорослих із Ph+ ALL на один цикл іматинібу плюс хіміотерапія низької інтенсивності (вінкристин) проти одного циклу ІТК плюс хіміотерапія високої інтенсивності (гіпер-CVAD; гіперфракціонований циклофосфамід, вінкристин, доксорубіцин, дексаметазон).
Усі пацієнти, хто досяг великої молекулярної відповіді, перейшли до алогенної або аутологічної трансплантації стовбурових клітин.
У порівнянні з гіпер-CVAD терапія низької інтенсивності була пов’язана з вищим рівнем CR (98 проти 91 %) і меншою кількістю ранніх смертей (1 проти 7 %). Рівень MMolR був подібним в обох групах (66 проти 64 %), і не було різниці в п’ятирічних показниках безрецидивної або загальної виживаності (37 і 46 % відповідно). При терапії низької інтенсивності спостерігалося менше побічних ефектів ≥3 ступеня, включаючи нейтропенію (6 проти 14 % відповідно), тромбоцитопенію (0 проти 3 %), інфекційні події (37 проти 58 %) та іншу токсичність (41 проти 46 %).
Немає єдиної думки щодо визначення схем хіміотерапії низької, середньої та високої інтенсивності. Важливо, що оскільки багато пацієнтів з Ph+ ALL є старшими та/або мають значні супутні захворювання, навіть схеми низької інтенсивності можуть бути пов’язані зі значними побічними ефектами.
Нижче наведено приклади схем на основі хіміотерапії, які були поєднані з ІТК для Ph+ ALL:
Низька інтенсивність – схеми низької інтенсивності такими, що включають лише один немієлосупресивний хіміотерапевтичний засіб (наприклад, вінкристин) плюс глюкокортикоїд. Ці схеми придатні для пацієнтів з будь-яким рівнем фізичної підготовки, але потрібна обережність для хворих із слабкістю або похилого віку, оскільки вінкристин може спричинити кишкову непрохідність або обстипацію у літніх пацієнтів, особливо якщо вони прикуті до ліжка. Вибір протоколу низької інтенсивності залежить від закладу, але EWALL-PH-01 є прикладом режиму низької інтенсивності:
Дексаметазон (10 мг на день протягом п’яти днів), потім дазатиніб (100 мг перорально на день), вінкристин (2 мг внутрішньовенно дні 1, 8, 15, 22) і дексаметазон 40 мг протягом двох днів. Для пацієнтів старше 70 років дозу вінкристину зменшують до 1 мг, а дозу дексаметазону – до 20 мг на добу протягом двох днів. Негематологічні побічні ефекти в основному були інфекціями, набряками та периферичною нейропатією.
Лікування 71 пацієнта (середній вік 69 років) EWALL-PH-01 було пов’язано з 96 % CR та 65 % великої молекулярної відповіді. Коефіцієнти п’ятирічної загальної і безрецидивної виживаності становили 36 і 28 % відповідно; лише сім пацієнтів пройшли алогенну трансплантацію стовбурових клітин Серед пацієнтів, у яких зрештою стався рецидив, мутація BCR-ABL1 T315I була виявлена у трьох чвертей обстежених. Побічні ефекти ≥3 ступеня включали інфекції (37 %) та інші негематологічні події (41 %).
Помірна інтенсивність – більшість мультиагентних схем і/або схем, що містять антрацикліни, є хіміотерапією помірної інтенсивності. Такі схеми придатні для придатних за станом здоров’я пацієнтів і для окремих пацієнтів, які не придатні за медичними показаннями, але не слабкі.
●CALGB 10701 – Схема CALGB 10701 починається з циклу дазатинібу плюс дексаметазону, після чого системно та інтратекально вводиться метотрексат , а для пацієнтів із >20 % лімфобластів у кістковому мозку – вінкристин і даунорубіцин.
Попередні результати, представлені в абстрактній формі, повідомляють про 45-місячну медіану ЗВ, 55% трирічну ЗВ і 43% трирічну безрецидивну виживаність серед 64 пацієнтів, які спостерігалися протягом чотирьох років.
●Міні-hyperCVD (варіант гіпер-CVAD, який використовує знижені дози хіміотерапії та не включає антрациклін) та інші схеми помірної інтенсивності повідомляли про подібні результати.
Високоінтенсивна – високоінтенсивні режими хіміотерапії підходять лише для здорових пацієнтів. Такі режими можуть досягти стійкої молекулярної відповіді, але вони пов’язані зі значною захворюваністю, пов’язаною з лікуванням, і можливою смертністю.
Hyper-CVAD включає вісім 21-денних циклів гіперфракціонованого циклофосфаміду, MESNA, доксорубіцину, вінкристину, дексаметазону, які чергуються з метотрексатом, цитарабіном і лейковорином, а також інгібітором ІТК (наприклад, понатініб, дазатиніб або іматиніб).
Лікування 76 пацієнтів (середній вік 47 років) понатінібом і гіпер-CVAD було пов’язано зі 100 % CR, 97 % MMolR, 73 % molCR і 70 % трирічної EFS.
Найпоширенішими побічними явищами ≥3 ступеня були інфекція (86 %), підвищення трансаміназ (32 %), підвищення білірубіну (17 %), панкреатит (17 %), гіпертонія (16 %), кровотеча (13 %) і висип ( 12 %); шість пацієнтів померли під час прийому дослідного препарату, у тому числі троє від інфекції, один від крововиливу та двоє від інфаркту міокарда.
РЕЖИМИ ДЛЯ Ph-ПОЗИТИВНИХ B-ALL
AYA пацієнти
| Інші рекомендовані режими |
| Схема EsPhALL: TKI + основа схеми Берлін-Франкфурт-Мюнстер (циклофосфамід, вінкристин, даунорубіцин, дексаметазон, цитарабін, метотрексат, пегаспаргаза та преднізон) • TKI + гіпер-CVAD (гіперфракціонований циклофосфамід, вінкристин, доксорубіцин і дексаметазон), чергуючи з високими дозами метотрексату і цитарабіну • ТКІ + мультиагентна хіміотерапія • ТКІ+ кортикостероїди • TKI • ТКІ + вінкристин + дексаметазон • Схема CALGB 10701: TKI + мультиагентна хіміотерапія (дексаметазон, вінкристин, даунорубіцин, метотрексат, етопозид та цитарабін) • Блінатумомаб ± ІТК |
Дорослі пацієнти (<65 років і без істотних супутніх захворювань)
| Інші рекомендовані режими |
| TKI + гіпер-CVAD (гіперфракціонований циклофосфамід, вінкристин, доксорубіцин і дексаметазон), чергуючи з високою дозою метотрексату і цитарабіну • ТКІ + мультиагентна хіміотерапія • ТКІ + кортикостероїди • TKI • ТКІ + вінкристин + дексаметазон • Схема CALGB 10701: TKI + мультиагентна хіміотерапія (дексаметазон, вінкристин, даунорубіцин, метотрексат, етопозид та цитарабін) • Блінатумомаб ± ІТК |
Лікування вперше діагностованого гострого лімфобластного лейкозу (ГЛЛ) із позитивною філадельфійською хромосомою (Ph+) у дорослих.
| Індукційна терапія |
| Інгібітор тирозинкінази BCR-ABL (TKI) плюс одна з наступних комбінованих схем хіміотерапії: – Рак і лейкемія Група B (дослідження CALGB 8811 або 9111) Схема ALL – Гіперфракціонований циклофосфамід, вінкристин, доксорубіцин і дексаметазон (Hyper-CVAD) чергування з високими дозами метотрексату та цитарабіну – Французька схема GRAALL-2003 – Німецькі протоколи GMALL 06/99 та 7/03 Комбінація ІТК з кортикостероїдом або без нього здатна призвести до повної ремісії при Ph+ ALL, які раніше не лікувалися, вона має низьку токсичність . Ця комбінація не порівнювалася безпосередньо з ІТК плюс хіміотерапія. Якщо початкові дані підтвердяться подальшими дослідженнями, комбінація ІТК і стероїдів може стати відмінним варіантом для пацієнтів, наприклад літніх людей, які не можуть переносити комбіновану хіміотерапію. |
| Консолідація |
| Перевага віддається алогенній трансплантації гемопоетичних клітин у першій повній ремісії. Якщо пацієнт не є кандидатом на трансплантацію, слід провести консолідаційну хіміотерапію, яка включає BCR-ABL TKI. |
| Підтримуюча терапія |
| BCR-ABL TKI протягом двох років після алогенної трансплантації гемопоетичних клітин або на невизначений термін, якщо трансплантація не виконана. |
| Спостереження |
| Оцінка кісткового мозку та тестування на мінімальну залишкову хворобу з транскриптами BCR-ABL за допомогою кількісної полімеразної ланцюгової реакції в реальному часі. Примітка: обраний BCR-ABL TKI має відповідати тирозинкіназі BCR-ABL, яка використовується в конкретному протоколі лікування. |
Моніторинг та оцінка відповіді
Моніторинг під час індукційної терапії.
Пацієнти, які проходять індукційну терапію, потребують щоденного клінічного та лабораторного обстеження для лікування цитопеній, метаболічних порушень та оцінки ускладнень лейкемії.
Більшість пацієнтів потребують госпіталізації з підтримкою препаратів крові під час терапії індукції ремісії. Іноді окремі пацієнти можуть лікуватися амбулаторно. Щоденне лабораторне обстеження повинно включати загальний аналіз крові та біохімічний аналіз функції нирок, глюкози та електролітів. Рівні кальцію, фосфору та сечової кислоти слід контролювати до нормалізації, а аналізи функції печінки слід оцінювати принаймні щотижня.
Підтримуюча терапія є критично важливим компонентом індукційної терапії, включаючи лікування цитопеній, інфекцій, лізису пухлини та інших ускладнень, які супроводжують лікування гострого лейкозу.
Оцінка відповіді – Основною метою індукційної терапії є досягнення початкової повної гематологічної ремісії (ПВ). Подальше лікування визначається тим, чи досяг пацієнт CR проти меншої відповіді (тобто рефрактерного захворювання).
Дослідження кісткового мозку зазвичай проводиться, коли абсолютна кількість нейтрофілів становить >1000/мікрол, а кількість тромбоцитів >100 000/мікрол після індукційної терапії.
Для точної оцінки клітин кісткового мозку необхідна біопсія.
- Мікроскопія – гематологічний CR визначається як <5 % бластів у кістковому мозку та крові та відновлення нормального кровотворення (>25 % клітинності та нормальних показників периферичної крові).
- Цитогенетика – кістковий мозок та/або периферичну кров слід досліджувати на персистенцію філадельфійської хромосоми за допомогою хромосомних смуг або флуоресцентної гібридизації in situ (FISH), щоб задокументувати ступінь повної цитогенетичної ремісії (CCyR). Важливість досягнення CCyR була продемонстрована в міжнародному дослідженні ALL, яке включало як Ph+ ALL, так і Ph-негативний ALL; порівняно з пацієнтами, які не досягли CCyR, загальна виживаність (ЗВ) була кращою для пацієнтів, які досягли CCyR (45 проти 5 %, відповідно).
- Вимірна залишкова хвороба (MRD) – MRD оцінюється за допомогою кількісної полімеразної ланцюгової реакції (RQ-PCR) для BCR-ABL1. MRD для Ph+ ALL обговорюється нижче.
Подальше лікування визначається відповіддю на індукційну терапію:
- Повна ремісія – для пацієнтів, які досягли CR, лікування після ремісії включає інгібітор тирозинкінази (TKI) плюс консолідаційну терапію (наприклад, імунотерапію, хіміотерапію та/або трансплантацію гемопоетичних клітин) з наступною підтримуючою терапією.
- Молекулярна ремісія – повна морфологічна ремісія плюс невизначуваний BCR-ABL1 за допомогою кількісної полімеразної ланцюгової реакції зворотної транскриптази (RT-PCR).
- Рефрактерна хвороба – пацієнти, які не досягають CR за допомогою індукційної терапії, вважаються такими, що мають рефрактерну хворобу.
Оцінка відповіді на лікування
| Відповідь | Показники |
| Повна ремісія (CR) | Відсутність циркулюючих лімфобластів або екстрамедулярного захворювання Відсутність лімфаденопатії, спленомегалії, інфільтрації шкіри/ясен/ураження яєчка/ураження ЦНС Трилінійний гемопоез і <5% бластів Абсолютна кількість нейтрофілів (ANC) ≥1000/мікрол Тромбоцити ≥100 000/мікрол |
| CR з частковим гематологічним відновленням (CRh) | Відповідає всім критеріям CR, за винятком часткового відновлення периферії аналіз крові (тромбоцити ≥50 000/мікрол і ANC ≥500/мікрол) |
| CR з неповним гематологічним відновленням (CRi) | Відповідає всім критеріям CR, за винятком без відновлення кількості тромбоцитів або без відновлення ANC (тромбоцити <100 000/мікрол і ANC ≥1000/мікрол або тромбоцити ≥100000/мікрол та ANC <1000/мікрол) |
| Морфологічний стан без лейкемії (MLFS): | Бласти <5% і відсутність екстрамедулярного лейкозу ANC <500/мікрол і тромбоцити <50/мікрол Клітинність кісткового мозку становить ≥10%, щонайменше 200 клітин |
| Апластичний кістковий мозок | Усі критерії для MLFS відповідають, але з клітинністю <10% та/або аспірат з <200 клітинами, які можна перерахувати |
| Рефрактерна хвороба | Неможливість досягти CR наприкінці індукції |
| Прогресуюча хвороба (ПЗ) | Поява циркулюючих лейкозних бластів або принаймні збільшення 25% в абсолютній кількості циркулюючих або бластів кісткового мозку або розвиток екстрамедулярного захворювання. |
| Рецидив хвороби | Повторна поява бластів у крові чи кістковому мозку (>5%) або будь-яке екстрамедулярне ураження після CR. |
Моніторинг MRD
Виявлення та моніторинг MRD є важливим аспектом лікування дорослих із ГЛЛ з позитивною (Ph+) і негативною (Ph-) філадельфійською хромосомою.
Вплив на прогноз – залежно від використовуваного методу, до 80 % дорослих з ГЛЛ матимуть виявлення MRD одразу після завершення індукційної терапії.
Численні дослідження показали, що пацієнти з виявленою MRD мають значно вищий рівень рецидивів.
MRD найчастіше виявляється за допомогою методів полімеразної ланцюгової реакції (ПЛР) і багатобарвної проточної цитометрії. ПЛР зі зворотною транскриптазою (RT-PCR) можна використовувати для виявлення специфічної для лейкемії злитої мРНК, такої як BCR::ABL, отриманої в результаті t(9;22). Досліджуються більш надійні методи з використанням мультиплексної ПЛР з глибоким секвенуванням і проточною цитометрією нового покоління.
Виявлення пацієнтів із стійкою MRD, які мають найвищий ризик рецидиву, може уможливити терапію після ремісії новими препаратами, такими як інотузумаб, озогаміцин, блінатумомаб і CD19-спрямовані химерні антигенні рецепторні Т-клітини (CAR-T), усі з яких були показані бути ефективним у досягненні негативного MRD, навіть у пацієнтів з рецидивом або рефрактерним захворюванням.
Ph+ ALL – злитий ген BCR::ABL, пов’язаний із філадельфійською (Ph) хромосомою, t(9;22), який зустрічається приблизно у 30 % дорослих із ГЛЛ, є маркером поганого прогнозу. Такі пацієнти регулярно лікуються алогенною трансплантацією гемопоетичних клітин (HCT) після індукції ремісії.
RT-PCR можна використовувати для виявлення та моніторингу злитого гена BCR::ABL. Існує два основних варіанти гена злиття. Приблизно 70 % пацієнтів мають підтип p190. Підтип p210 (присутній у переважної більшості пацієнтів з хронічним мієлоїдним лейкозом) становить від 25 до 30 % випадків.
Виявлення MRD за допомогою RT-PCR аналізу BCR::ABL передбачає ранній рецидив. Як приклад, в одному дослідженні повідомлено, що статус MRD на момент першої повної ремісії не був прогностичним, але статус MRD через три місяці та після прогнозував виживання. Невідомо, чи можна цих пацієнтів вилікувати без алогенної HCT.
Декілька досліджень статусу MRD після алогенної або аутологічної HCT у пацієнтів з ГЛЛ, позитивною за філадельфійською хромосомою, свідчать про те, що позитивний MRD може ідентифікувати пацієнтів, які мають ймовірність рецидиву. У найбільшій серії відносний ризик рецидиву був значно вищим для пацієнтів із виявленим транскриптом BCR::ABL після HCT порівняно з пацієнтами без виявленого BCR::ABL (відносний ризик 5,7, p = 0,025) . Середній час від виявлення позитивного результату ПЛР до клінічного рецидиву становив 94 дні.
Терміни оцінки MRD:
- Після завершення первинної індукції.
- Кінець консолідації.
- Додаткове визначення MRD – слід орієнтуватися на використовуваний режим ПХТ.
- Частота моніторингу може бути збільшена у пацієнтів із молекулярним рецидивом або стійким низьким рівнем залишкової хвороби.
- Для деяких методів базовий зразок (тобто перед лікуванням) необхідний для характеристики лейкемічного клону для подальшої оцінки MRD.
Зареєструйтеся на нашому сайті прямо зараз, щоб мати доступ до більшої кількості навчальних матеріалів!
Підписатися на наші сторінки:
Список джерел:
- NCCN clinical guidelines
- Yan L., Ping N., Zhu M., Sun A., Xue Y., Ruan C., Drexler H.G., MacLeod R.A.F., Wu D., Chen S. Clinical, immunophenotypic, cytogenetic, and molecular genetic features in 117 adult patients with mixed-phenotype acute leukemia defined by WHO-2008 classification. Haematologica. 2012;97:1708–1712. doi: 10.3324/haematol.2012.064485. PMC free article PubMed
- Bene M.C., Castoldi G., Knapp W., Ludwig W.D., Matutes E., Orfao A., van’t Veer M.B. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL) Leukemia. 1995;9:1783–1786. PubMed Google Scholar
- Borowitz M.J., Harris N.L., Porwit A., Matutes E. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. World Healh Organization Classification of Tumours; Lyon, France: 2008. Acute leukemias of ambiguous lineage. Google Scholar
- Swerdlow S.H.C.E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised. 4th ed. WHO Classification of Tumours; Lyon, France: 2017. Google Scholar
- Charles N.J., Boyer D.F. Mixed-Phenotype Acute Leukemia: Diagnostic Criteria and Pitfalls. Arch. Pathol. Lab. Med. 2017;141:1462–1468. doi: 10.5858/arpa.2017-0218-RA. PubMed
- Mi X., Griffin G., Lee W., Patel S., Ohgami R., Ok C.Y., Wang S., Geyer J.T., Xiao W., Roshal M., et al. Genomic and clinical characterization of B/T mixed phenotype acute leukemia reveals recurrent features and T-ALL like mutations. Am. J. Hematol. 2018;93:1358–1367. doi: 10.1002/ajh.25256. PMC free article PubMed
- Béné M.C., Lacombe F., Porwit A. Unsupervised flow cytometry analysis in hematological malignancies: A new paradigm. Int. J. Lab. Hematol. 2021;43:54–64. doi: 10.1111/ijlh.13548. PubMed
- Porwit A., Béné M.C. Acute leukemias of ambiguous origin. Am. J. Clin. Pathol. 2015;144:361–376. doi: 10.1309/AJCPSTU55DRQEGTE. PubMed
- Austin G.E., Alvarado C.S., Austin E.D., Hakami N., Zhao W.-G., Chauvenet A., Borowitz M.J., Carroll A.J. Prevalence of Myeloperoxidase Gene Expression in Infant Acute Lymphocytic Leukemia. Am. J. Clin. Pathol. 1998;110:575–581. doi: 10.1093/ajcp/110.5.575. PubMed
- Weinberg O.K., Chisholm K.M., Ok C.Y., Fedoriw Y., Grzywacz B., Kurzer J.H., Mason E.F., Moser K.A., Bhattacharya S., Xu M., et al. Clinical, immunophenotypic and genomic findings of NK lymphoblastic leukemia: A study from the Bone Marrow Pathology Group. Mod. Pathol. 2021;34:1358–1366. doi: 10.1038/s41379-021-00739-4. PubMed
- Quesada A.E., Hu Z., Routbort M.J., Patel K.P., Luthra R., Loghavi S., Zuo Z., Yin C.C., Kanagal-Shamanna R., Wang S.A., et al. Mixed phenotype acute leukemia contains heterogeneous genetic mutations by next-generation sequencing. Oncotarget. 2018;9:8441–8449. doi: 10.18632/oncotarget.23878. PMC free article PubMed
- Alexander T.B., Gu Z., Iacobucci I., Dickerson K., Choi J.K., Xu B., Payne-Turner D., Yoshihara H., Loh M.L., Horan J., et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature. 2018;562:373–379. doi: 10.1038/s41586-018-0436-0. PMC free article PubMed
- Zheng C., Wu J., Liu X., Ding K., Cai X., Zhu W. What is the optimal treatment for biphenotypic acute leukemia? Haematologica. 2009;94:1778–1780. doi: 10.3324/haematol.2009.014829. PMC free article PubMed
- Oberley M.J., Raikar S.S., Malvar J., Wertheim G., Seif A.E., Guinipero T., Sposto R., Rabin K.R., Punia J.N., Orgel E. Minimal residual disease risk-stratification in pediatric mixed phenotype acute leukemia: Results of a multi-center cohort study. Blood. 2018;132:558–1558. doi: 10.1182/blood-2018-99-113606. CrossRef Google Scholar
- Qasrawi A., Ramlal R., Munker R., Hildebrandt G.C. Prognostic impact of Philadelphia chromosome in mixed phenotype acute leukemia ( MPAL ): A cancer registry analysis on real-world outcome. Am. J. Hematol. 2020;95:1015–1021. doi: 10.1002/ajh.25873. PubMed
- Oberley M.J., Raikar S., Wertheim G.B., Malvar J., Sposto R., Rabin K.R., Punia J.N., Seif A.E., Cahen V.C., Schore R.J., et al. Significance of minimal residual disease in pediatric mixed phenotype acute leukemia: A multicenter cohort study. Leukemia. 2020;34:1741–1750. doi: 10.1038/s41375-020-0741-0. PMC free article PubMed
- Wolach O., Amitai I., DeAngelo D. Current challenges and opportunities in treating adult patients with Philadelphia-negative acute lymphoblastic leukaemia. Br. J. Haematol. 2017;179:705–723. doi: 10.1111/bjh.14916. PubMed
- Abaza Y., Fathi A.T. Monoclonal Antibodies in Acute Myeloid Leukemia-Are We There Yet? Cancer J. 2022;28:37–42. doi: 10.1097/PPO.0000000000000577. PubMed
- Munker R., Labopin M., Esteve J., Schmid C., Mohty M., Nagler A. Mixed phenotype acute leukemia: Outcomes with allogeneic stem cell transplantation. A retrospective study from the Acute Leukemia Working Party of the EBMT. Haematologica. 2017;102:2134–2140. doi: 10.3324/haematol.2017.174441. PMC free article PubMed
- Munker R., Brazauskas R., Wang H.L., de Lima M., Khoury H.J., Gale R.P., Maziarz R.T., Sandmaier B.M., Weisdorf D., Saber W. Allogeneic Hematopoietic Cell Transplantation for Patients with Mixed Phenotype Acute Leukemia. Biol. Blood Marrow Transplant. 2016;22:1024–1029. doi: 10.1016/j.bbmt.2016.02.013. PMC free article PubMed
- Bennett JM, Catovsky D, Daniel MT, et al. French-American-British (FAB Cooperative Group) Proposals for the classification of the acute leukaemias. Br J Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. PubMed
- Jaffe ES, Harris NL, Stein H, et al. Introduction an overview of the classification of the lymphoid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of tumours of haematopoietic and lymphoid tissue. IARC; Lyon: 2008. pp. 158–166. Google Scholar
- Stein P, Peiper S, Butler D, et al. Granular acute lymphoblastic leukemia. Am J Clin Pathol. 1983;80:545. PubMed
- Kalina T, Flores-Montero J, van der Velden VH, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26:1986–2010. doi: 10.1038/leu.2012.122. PMC free article PubMed
- Coustan-Smith E, Behm FG, Sanchez J, et al. Immunological detection of minimal residual disease in children with acute lymphoblastic leukaemia. Lancet. 1998;351:550–554. doi: 10.1016/S0140-6736(97)10295-1. PubMed
- Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. PMC free article PubMed
- Schardt C, Ottmann OG, Hoelzer D, Ganser A. Acute lymphoblastic leukemia with the (4;11) translocation: combined cytogenetic, immunological and molecular genetic analyses. Leukemia. 1992;6:370–374. PubMed
- Faderl S, Albitar M. Insights into the biologic and molecular abnormalities in adult acute lymphocytic leukemia. Hematol Oncol Clin North Am. 2000;14:1267–1288. doi: 10.1016/S0889-8588(05)70186-6. PubMed
- Westbrook CA, Hooberman AL, Spino C, et al. Clinical significance of the BCR-ABL fusion gene in adult acute lymphoblastic leukemia: a Cancer and Leukemia Group B Study (8762) Blood. 1992;80:2983–2990. PubMed
- Ravandi F, Jorgensen JL, Thomas DA, et al. Detection of MRD may predict the outcome of patients with Philadelphia chromosome-positive ALL treated with tyrosine kinase inhibitors plus chemotherapy. Blood. 2013 Aug 15;122:1214–1221. doi: 10.1182/blood-2012-11-466482. PMC free article PubMed
- Marks DI, Moorman AV, Chilton L, et al. The clinical characteristics, therapy and outcome of 85 adults with acute lymphoblastic leukemia and t(4;11)(q21;q23)/MLL-AFF1 prospectively treated in the UKALLXII/ECOG2993 trial. Haematologica. 2013;98:945–952. doi: 10.3324/haematol.2012.081877. PMC free article PubMed
- Hof J, Krentz S, van Schewick, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–93. doi: 10.1200/JCO.2011.34.8144. PubMed
- Yanada M, Takeuchi J, Sugiura I, et al.; Japan Adult Leukemia Study Group . High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol. 2006;24(3):460-466. PubMed
- Bassan R, Rossi G, Pogliani EM, et al.. Chemotherapy-phased imatinib pulses improve long-term outcome of adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: Northern Italy Leukemia Group protocol 09/00. J Clin Oncol. 2010;28(22):3644-3652. PubMed
- Lilly MB, Ottmann OG, Shah NP, et al.. Dasatinib 140 mg once daily versus 70 mg twice daily in patients with Ph-positive acute lymphoblastic leukemia who failed imatinib: Results from a phase 3 study. Am J Hematol. 2010;85(3):164-170. PubMed
- Vignetti M, Fazi P, Cimino G, et al.. Imatinib plus steroids induces complete remissions and prolonged survival in elderly Philadelphia chromosome-positive patients with acute lymphoblastic leukemia without additional chemotherapy: results of the Gruppo Italiano Malattie Ematologiche dell’Adulto (GIMEMA) LAL0201-B protocol. Blood. 2007;109(9):3676-3678. PubMed
- Porkka K, Koskenvesa P, Lundán T, et al.. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112(4):1005-1012. PubMed
Консультування пацієнта з підозрою на порфірію – рекомендації ОСКІ
Початок консультації Вимийте руки та при необхідності надіньте ЗІЗ (засоби індивідуального захисту) Представтеся пацієнту, вказуючи…

Консультування пацієнта при підозрі на гостру лімфобластну лейкемію – рекомендації OSCE
Початок консультації Вимийте руки та одягніть ЗІЗ, якщо це необхідно Представтесь пацієнту, вказавши своє ім’я…

Гостра мієлоїдна лейкемія
Гостра мієлоїдна лейкемія (ГМЛ) включає гетерогенну групу агресивних ракових клітин крові, які виникають внаслідок клональної…

Консультування пацієнта при підозрі на гостру мієлоїдну лейкемію – рекомендації ОСКІ
Початок консультації Вимийте руки та одягніть ЗІЗ, якщо це необхідно Представтесь пацієнту, вказавши своє ім’я…

Консультування пацієнта з хронічною мієлоїдною лейкемією – рекомендації OSCE
Автори: Юлія Малишева, Дмитро Гамов, Наталія Лопіна Початок консультації Вимийте руки та одягніть ЗІЗ,…
Хронічна мієлоїдна лейкемія
Хронічна мієлоїдна лейкемія – це мієлопроліферативне новоутворення, що характеризується нерегульованою продукцією та неконтрольованою проліферацією зрілих…