Множинна мієлома
Автори: Юлія Малишева, Дмитро Гамов
Множинна мієлома (ММ) – це злоякісне новоутворення з плазматичних клітин, які накопичуються в кістковому мозку, що призводить до руйнування кісток і кістковомозкової недостатності. Найчастіше ММ діагностується в людей віком від 65 до 74 років, а медіана віку становить 69 років.
Захворюваність та епідеміологія
Множинна мієлома (ММ) становить 1% усіх ракових захворювань і 10% усіх гематологічних злоякісних новоутворень. Захворюваність в Європі становить 4,5–6,0/100 000/рік із середнім віком на момент встановлення діагнозу 72 роки. Смертність становить 4,1/100 000/рік . Майже у всіх хворих ММ розвивається з безсимптомного передракового захворювання, що називається моноклональною гаммапатією невизначеного значення (MGUS). MGUS прогресує до MM зі швидкістю 1% на рік. У деяких пацієнтів проміжна безсимптомна, але більше прогресуюча передракова стадія, яка називається тліючою (або млявою).
Клінічні симптоми
Клінічні симптоми виникають внаслідок проліферації пухлинних плазмоцитів та секреції ними моноклональних (патологічних) білків і цитокінів:
- загальні cимптоми — слабкість і втрата маси тіла;
- оссалгія (найчастіший прояв) — локалізується в поперековому відділі хребта, кістках тазу, ребрах, рідше в черепі та трубчастих кістках, зумовлена остеолітичними змінами та патологічними переломами кісток (напр., компресійними переломами хребців);
- неврологічні симптоми — внаслідок компресії або ушкодження спинного мозку, спинномозкових корінців або черепних нервів патологічними переломами (напр. хребців) або безпосередньо пухлиною: найчастіше радикулопатія, іноді парези, паралічі кінцівок, нетримання сечі або калу; сенсорна або сенсомоторна периферична нейропатія, симетрична і дистальна, рідко на момент діагностування захворювання, частіша у хворих із супутнім амілоїдозом легких ланцюгів імуноглобулінів та при POEMS-синдромі, а також у хворих, які отримують нейротоксичні ЛЗ (талідомід, бортезоміб);
- симптоми анемії (≈70 %);
- прояви гіперкальціємії та її наслідків;
- повторні бактеріальні інфекції дихальної та сечовидільної систем, а також вірусні інфекції (грип, оперізуючий герпес);
- симптоми ниркової недостатності — у ≈30 % хворих на момент встановлення діагнозу ММ; найчастіше це т. зв. циліндрова нефропатія (тубуло-інтерстиціальний нефрит, спричинений інтратубулярними циліндрами, які сформувались із легких ланцюгів у сечі);
- прояви синдрому підвищеної в’язкості крові (у <10 % хворих): найчастіше геморагічний діатез (кровотечі з носа і ясен, пурпура), погіршення гостроти зору, прояви з боку ЦНС (біль голови, раптова глухота, запаморочення, атаксія, ністагм, розлади свідомості), загострення серцевої недостатності;
- рідше — екстрамедулярні плазмоцитоми, симптоми супутнього AL-амілоїдозу, гепатомегалія, збільшення периферичних лімфовузлів та селезінки, сидром де Тоні-Дебре-Фанконі.
Типовий перебіг
У приблизно 10–15 % хворих легкий перебіг (безсимптомна мієлома). У більшості випадків захворювання прогресує або рецидивує після наступних ліній лікування.
Діагностика та патологія/молекулярна біологія
Діагностика ММ повинна ґрунтуватися на наступних тестах:
- Виявлення та оцінка моноклональної (М) складової шляхом електрофорезу білків сироватки та/або сечі (концентрат 24-годинного збору сечі); нефелометрична кількісна оцінка IgG, імуноглобуліни IgA і IgM; характеристика важких і світлові ланцюги шляхом імунофіксації; і вимірювання світлового ланцюга без сироватки (FLC).
- Оцінка інфільтрації плазматичних клітин кісткового мозку: аспірація кісткового мозку та/або біопсія є стандартними варіантами оцінки кількості та характеристик плазматичних клітин у кістковому мозку. Крім того, зразок кісткового мозку слід використовувати для досліджень цитогенетичної/флуоресцентної гібридизації in situ (FISH) на імунологічно визнаних або відсортованих плазматичних клітинах, і він також може бути використаний для імунофенотипових та молекулярних досліджень.
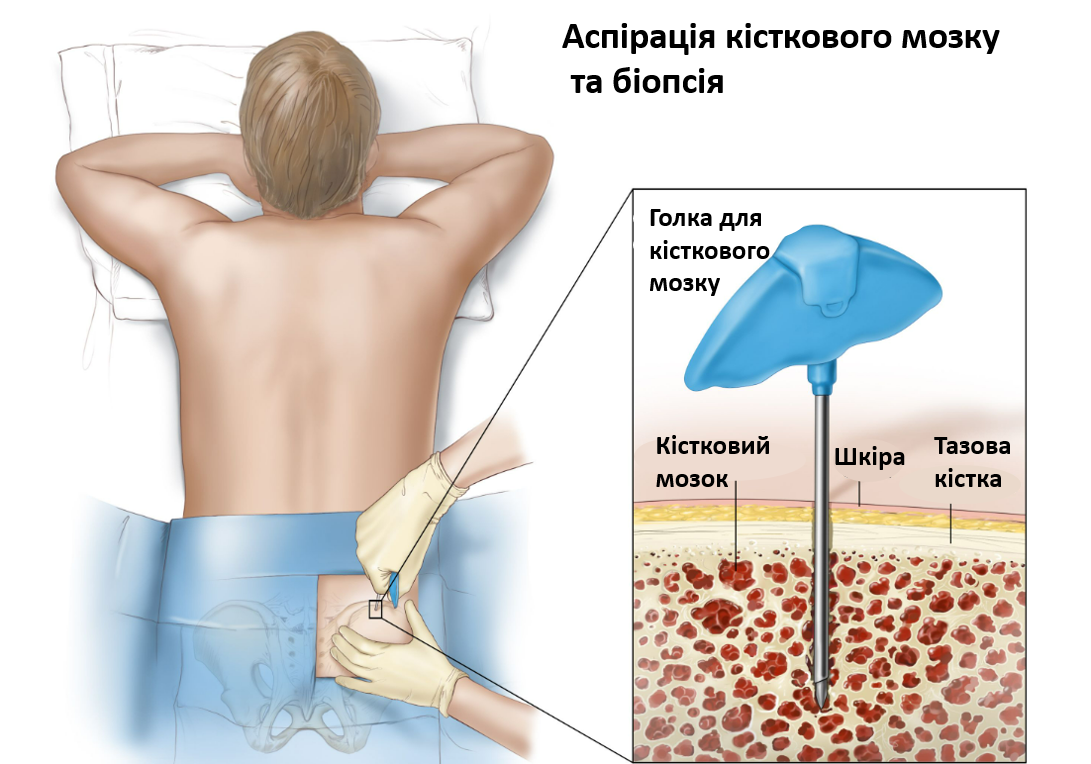
- Оцінка літичних уражень кісток: низькодозова комп’ютерна томографія (WBLD-CT) всього тіла є новим стандартом для діагностики літичної хвороби. Звичайна рентгенографія також може використовуватися, якщо WBLD-CT недоступний. Магнітно резонансна візуалізація (МРТ) надає більш детальну інформацію і рекомендується щоразу, коли підозрюється компресія спинного мозку.
Щоб відрізнити симптомну й безсимптомну ММ, необхідні такі лабораторні дослідження на вихідному рівні: загальний аналіз крові (ЗАК) із лейкоцитарною формулою та підрахунком тромбоцитів. - Дослідження мазка периферичної крові.
- Азот сечовини крові (АСК).
- Креатинін у сироватці крові.
- Кліренс креатиніну (розрахований або виміряний безпосередньо).
- Електроліти в сироватці крові; печінкові проби; кальцій у сироватці крові; альбумін; лактатдегідрогеназа (ЛДГ).
- Бета2 мікроглобулін.
- Дослідження мазка периферичної крові може показати аномальний розподіл еритроцитів, наприклад утворення «монетних стовпчиків» (тобто склеювання еритроцитів подібно до монетних стовпчиків) через підвищений рівень сироваткових білків.
- Підвищення АСК і креатиніну вказує на зниження функції нирок, водночас рівні ЛДГ та бета-2- мікроглобуліну відображають характеристики пухлинних клітин.
- Аналіз сироватки включає кількісне визначення рівня імуноглобулінів (IgG, IgA та IgM); електрофорез білків сироватки (serum protein electrophoresis, SPEP) для кількісного визначення моноклонального білка; електрофорез з імунофіксацією сироватки (serum immunofixation electrophoresis, SIFE) для отримання більш конкретної інформації про тип присутнього М-білка.
- Оцінка змін рівня різних білків, зокрема М-білка, допомагає відстежити прогресування захворювання та відповідь на лікування. Аналіз сечі в межах початкової діагностики включає оцінку добової сечі на загальний білок, електрофорез білків сечі (urine protein electrophoresis, UPEP) та електрофорез із імунофіксацією сечі (urine immunofixation electrophoresis, UIFE).
- Аналіз на вільні легкі ланцюги: аналіз сироватки на вільні легкі ланцюги (free light-chain, FLC) разом з аналізами сироватки (SPEP та SIFE) забезпечує високу чутливість під час скринінгу на ММ і споріднені плазмоклітинні захворювання . Він також допомагає в прогнозуванні моноклональної гамапатії невизначеного генезу (МГНГ), «тліючої» мієломи, активної ММ, амілоїдозу легких ланцюгів імуноглобулінів і солітарної плазмоцитоми . Аналіз сироватки на FLC також дає змогу проводити кількісний моніторинг пацієнтів з амілоїдозом легких ланцюгів і мієломою легких ланцюгів.
Окрім усього переліченого вище, співвідношення вільних легких ланцюгів (FLC ratio, FLCr) необхідне для документування строгої повної відповіді (СПВ) відповідно до Єдиних критеріїв відповіді Міжнародної робочої групи з мієломи (International Myeloma Working Group, IMWG).
Після кількісного визначення М-білка важливо використовувати той самий аналіз для серійних досліджень, щоб забезпечити точне відносне кількісне визначення.
Оцінка кісткового мозку
Для діагностики ММ основним критерієм є відсоток клональних плазматичних клітин кісткового мозку (≥ 10 %). Відсоток плазматичних клітин у кістковому мозку оцінюється шляхом односторонньої аспірації та біопсії кісткового мозку. Для підтвердження наявності моноклональних плазматичних клітин і для більш точної кількісної оцінки ураження плазматичних клітин можуть бути використані імуногістохімічний аналіз та/або проточна цитометрія. Цитоплазма аномальних плазматичних клітин містить легкі ланцюги каппа або лямбда, і переважання плазматичних клітини, які експресують той чи інший легкий ланцюг, вказує на клональність. Конкретні імунофенотипові профілі клітин мієломи можуть мати прогностичне значення.
Плазмоцити в мазку кісткового мозку
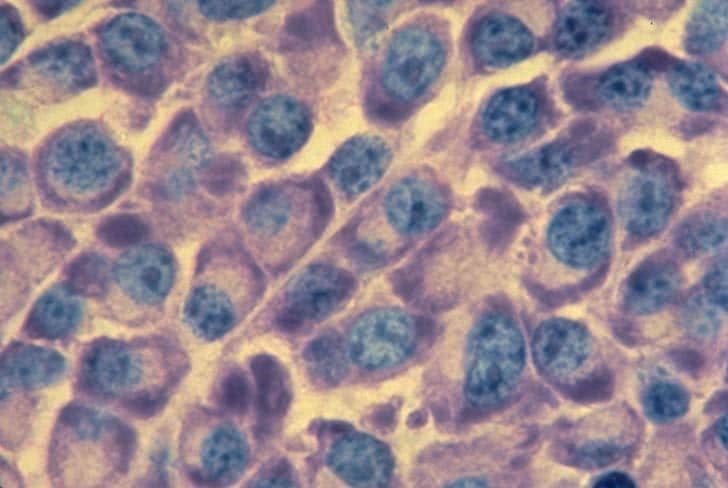
Існує кілька видів плазмоцитарних новоутворень. Усі ці захворювання пов’язані з моноклональним (або мієломним) білком (М-білком). Вони включають моноклональну гаммапатію невизначеного значення (MGUS), ізольовану плазмоцитому кістки, екстрамедулярну плазмоцитому та множинну мієлому.
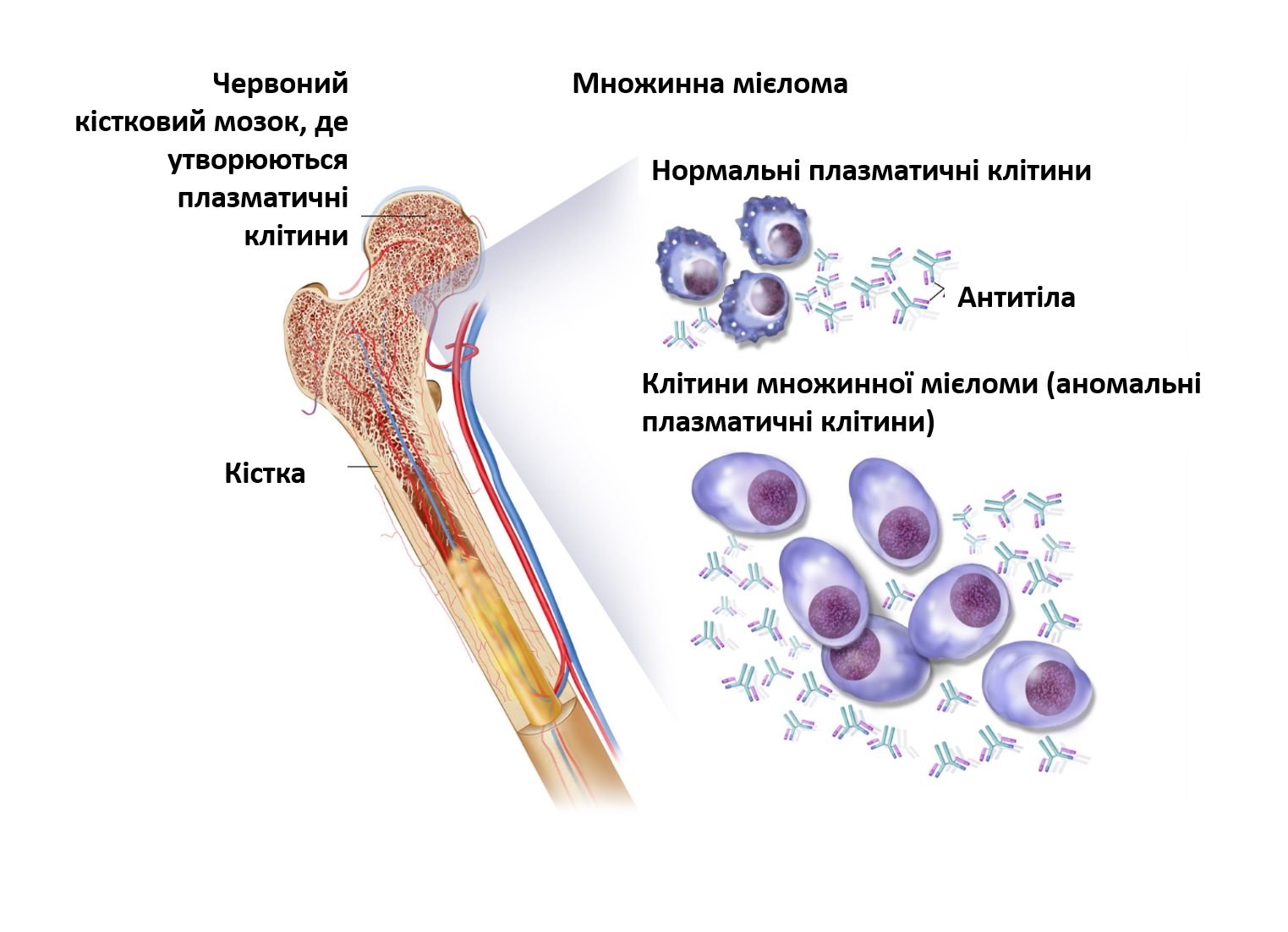
Важливо відрізняти ММ від інших плазмоклітинних новоутворень / гематологічних захворювань, щоб правильно визначити прогноз і призначити відповідне лікування.
| Плазматична неоплазма | Тип протеїну M | Патологія | Клінічна картина |
| Ig = імуноглобулін; MGUS = моноклональна гаммопатія невизначеного значення. | |||
| MGUS | IgG каппа або лямбда; або IgA каппа або лямбда | <10% плазматичних клітин у кістковому мозку | Безсимптомна, з мінімальними ознаками захворювання (окрім наявності білка М) |
| Ізольована плазмоцитома кістки | IgG каппа або лямбда; або IgA каппа або гамма | Солітарне ураження кісток; <10% плазматичних клітин у кістковому мозку неураженої ділянки | Безсимптомна або симптоматична |
| Екстрамедулярна плазмоцитома | IgG каппа або лямбда; або IgA каппа або гамма | Солітарне ураження м’яких тканин; найчастіше виникає в носоглотці, мигдаликах або навколоносових пазухах | Безсимптомна або симптоматична |
| Множинна мієлома | IgG каппа або лямбда; або IgA каппа або гамма | Нерідко множинні ураження кісток | Симптоматична |
Моноклональна гамапатія невизначеного значення (MGUS)
Пацієнти з MGUS мають білок М у сироватці крові без ознак множинної мієломи, макроглобулінемії, амілоїдозу або лімфоми та мають менше 10% плазматичних клітин у кістковому мозку .Пацієнти з тліючою мієломою мають подібні характеристики, але можуть мати більше 10% плазматичних клітин у кістковому мозку.
Ці типи пацієнтів протікають безсимптомно і не потребують лікування. Пацієнти з MGUS і факторами ризику прогресування захворювання, однак, повинні перебувати під ретельним наглядом, оскільки вони мають більшу ймовірність розвитку мієломи (найчастіше), амілоїдозу, лімфоплазмоцитарної лімфоми або хронічної лімфолейкемії, і тоді може знадобитися терапія.
Практично всім випадкам множинної мієломи передує поступове підвищення рівня MGUS. Річний ризик прогресування MGUS до лімфоїдної або плазматичної злоякісної пухлини коливається від 0,5% до 1,0% у популяційних когортах.
Фактори ризику, які передбачають прогресування захворювання, включають наступне:
- Аномальне співвідношення сироватка-FLC;
- MGUS класу не IgG;
- Високий рівень сироваткового білка М (≥1,5 г/дл).
Моноклональні гаммопатії, які спричиняють пошкодження органів, зокрема нирок, серця або периферичних нервів, вимагають негайної терапії з використанням тих самих стратегій, які застосовуються для звичайних плазмоцитарних дискразій. Моноклональна гаммопатія, що спричиняє дисфункцію нирок — через пряме відкладення антитіл або амілоїдоз — називається моноклональною гаммопатією ниркового значення. Підвищення рівня креатиніну в сироватці крові, зниження швидкості клубочкової фільтрації та збільшення екскреції альбуміну з сечею – це всі параметри, які можуть вказувати на пошкодження нирок і оцінюються проспективно для пацієнтів із MGUS високого ризику. Хоча N-кінцевий промозковий натрійуретичний пептид є дуже чутливим маркером залучення амілоїду в серце, слід зазначити низьку специфічність. Ці додаткові тести включені разом з рівнем М-білка, рівнями FLC і співвідношенням FLC під час спостереження за пацієнтами з MGUS.
Ізольована плазмоцитома кістки
У пацієнта ізольована плазмоцитома кістки, якщо виявлено:
- Солітарне літичне ураження плазматичних клітин при огляді скелета у безсимптомного пацієнта.
- Дослідження кісткового мозку з неураженої ділянки містить менше 10% плазматичних клітин.
- МРТ може виявити непередбачувані ураження кісток, які не були виявлені на стандартних рентгенограмах. МРТ всього хребта і тазу може виявити інші кісткові ураження.
Екстрамедулярна плазмоцитома
У пацієнта екстрамедулярна плазмоцитома, якщо виявлено:
- Ізольовані плазматичні пухлини м’яких тканин, які найчастіше виникають у мигдаликах, носоглотці або навколоносових пазухах.
- Негативні результати рентгенівських знімків скелета та біопсії кісткового мозку.

Множинна мієлома
Множинна мієлома — це системна злоякісна пухлина плазматичних клітин, яка зазвичай вражає кілька ділянок у кістковому мозку та секретує повністю або частково моноклональне антитіло.
Клінічні результати
На підставі результатів клінічного та лабораторного обстеження пацієнти спочатку класифікуються як такі, що мають моноклональну гамапатію невизначеного генезу (МГНГ), солітарну плазмоцитому, «тліюче» (безсимптомне) захворювання або активне (симптомне) захворювання. Останнім часом пацієнтів із МГНГ, які мають системні ефекти, пов’язані з моноклональною гамапатією, класифікують як таких, що мають клінічно значущу моноклональну гамапатію або моноклональну гамапатію ниркового генезу, залежно від характеру ураження органів.
Критерії CRAB
Критерії CRAB, що дають змогу визначити ММ, включають:
- С (calcium levels) — підвищений рівень кальцію (більше 11,5 мг/дл);
- R (renal insufficiency) — ниркову недостатність (креатинін більше 2 мг/дл або кліренс креатиніну менше 40 мл/хв);
- A (anemia) — анемію (гемоглобін менше 10 г/дл) на 2 г/дл менше норми;
- B (bone lesions) — наявність уражень кісток.
IMWG також уточнила, що наявність одного або кількох остеолітичних вогнищ ураження, виявлених під час рентгенографії скелета, МРТ всього тіла або ПЕТ/КТ усього тіла з ФДГ відповідає критеріям ураження кісток.
Біомаркери, які визначають ММ, визначені критеріями IMWG SLiM (SLiM розшифровується як Sixty — шістдесят, Light chain ratio — співвідношення легких ланцюгів, MRI — МРТ), включають одне або кілька із зазначеного далі:
- шістдесят або більше відсотків клональних плазматичних клітин у кістковому мозку;
- співвідношення залучених / незалучених FLC дорівнює 100 або більше, водночас рівень вражених вільних легких ланцюгів дорівнює або перевищує 100 мг/л;
- або МРТ із більше ніж одним осередковим ураженням кісткового мозку (не остеолітичним).
Всі ці явища, що визначають мієлому, називаються SLiM-CRAB.
Критерії IMWG для пацієнтів із «тліючою» (безсимптомною) мієломою включають сироватковий М-білок (IgG або IgA) ≥ 30 г/л та/або клональні плазматичні клітини кісткового мозку від 10 % до 59 % та відсутність ознак CRAB, явищ, що визначають мієлому, або амілоїдозу. Оновлені діагностичні критерії ММ IMWG дають змогу розпочати терапію до початку враження органів-мішеней на основі специфічних біомаркерів, а також використовувати чутливі критерії візуалізації для діагностики ММ, включно з ПЕТ/КТ усього тіла з ФДГ і МРТ.
Пацієнтів з активною ММ можна класифікувати за допомогою Міжнародної системи стадіювання (International Staging System, ISS). ISS ґрунтується на легко одержуваних лабораторних показниках (сироватковий бета-2-мікроглобулін і сироватковий альбумін) і більш проста у використанні, ніж система стадіювання Дюрі — Сальмона, для пацієнтів із раніше не лікованою ММ.
Міжнародна система визначення стадії (ISS) для множинної мієломи
| Стадія | Критерії | Середня виживанність (міс.) |
| I-FISH = інтерфазна флуоресцентна гібридизація in situ; ЛДГ = лактатдегідрогеназа; R-ISS = переглянута міжнародна система стадіювання | ||
| І | Бета-2-мікроглобулін <3,5 мг/л і альбумін ≥3,5 г/дл | Не досягнуто |
| II | Не R-ISS I або III | 83 |
| III | Бета-2-мікроглобулін ≥5,5 мг/л і високий рівень LDH або хромосомні аномалії високого ризику за І-FISH (визначаються як наявність del(17p) та/або транслокації t(4;14) та/або транслокації t(14) ;16)) | 43 |
Система ISS була переглянута (R-ISS) і тепер охоплює бета-2-мікроглобулін та альбумін сироватки крові, а також прогностичну інформацію, отриману на основі рівня ЛДГ та хромосомних аномалій високого ризику (t(4;14), t(14;16), делеція 17p13), виявлених за допомогою FISH, та є кращим методом стадіювання.
Пацієнти з del(17p) та/або транслокацією t(4;14) та/або транслокацією t(14;16) належать до групи високого ризику. Пацієнти без хромосомних аномалій високого ризику належать до групи стандартного ризику.
Генетичні фактори та групи ризику
Новітніші клінічні дослідження розподіляють пацієнтів із множинною мієломою на так звані групи стандартного ризику, середнього ризику та високого ризику на основі генетичних аберацій, виявлених за допомогою I-FISH. Стратифікація, заснована на цитогенетичних результатах, була отримана в результаті ретроспективного аналізу та потребує проспективної перевірки. Зразки кісткового мозку надсилаються на цитогенетичний аналіз і аналіз FISH. Плазмоклітинна лейкемія має особливо поганий прогноз. В іншому випадку сприятливий прогноз гіперплоїдії перевершується супутньою несприятливою цитогенетикою.
Групи ризику множинної мієломи
| Група ризику | Цитогенетичні дані | Характеристика захворювання | Медіана виживання (рік) |
| FISH = флуоресцентна гібридизація in situ; Ig = імуноглобулін. | |||
| Хороший ризик | Має будь-які з наведених нижче цитогенетичних знахідок: відсутність несприятливих FISH або цитогенетики, гіпердиплоїдія, t(11;14) за FISH або (4) t(6;14) за FISH. | Ці пацієнти найчастіше мають захворювання, що експресує IgG каппа моноклональні гаммопатії та літичні ураження кісток. | 8–10 |
| Проміжний ризик | t(4;14) за допомогою FISH | Ці пацієнти часто мають IgA-лямбда-моноклональні гаммопатії та рідше захворювання кісток. | 5 |
| Високий ризик | Має будь-які з наступних цитогенетичних знахідок: del 17p за FISH, t(14;16) за FISH, t(4;14), t(14;20), цитогенетичний del 13, негіпердиплоїдія без несприятливих цитогенетичних знахідок, збільшення 1q або лейкоз плазмоцитів. | Ці пацієнти мають захворювання, що експресує IgA лямбда моноклональні гаммопатії (часто) та ускладнення, пов’язані з скелетом (рідше). | 2 |
Хоча ММ може бути морфологічно схожою, на генетичному й молекулярному рівнях було виявлено кілька підтипів захворювання. Дослідження кісткового мозку під час початкової діагностики мають включати аналіз хромосом методом флуоресцентної гібридизації in situ (fluorescence in situ hybridization, FISH), проведений на плазматичних клітинах, отриманих під час аспірації кісткового мозку. Додаткову інформацію можна отримати за допомогою метафазного цитогенетичного дослідження.
У пацієнтів із ММ були виявлені специфічні хромосомні аномалії, що включають транслокацію, делецію або ампліфікацію. Делеція 17p13 (локусу для гена-супресора пухлини, p53) призводить до втрати гетерозиготності гена TP53 та вважається ознакою високого ризику розвитку ММ10-12. Цей ризик достовірно підвищується за умови більш високої частки клітин мієломи з цією аномалією, а також мутації алелю, що залишився.
Інші хромосомні аберації високого ризику за ММ характеризуються структурними змінами, які включають специфічні перебудови за участю гена IGH (який кодує важкий ланцюг імуноглобуліну), розташованого в позиції 14q32. На підставі транслокацій 14q32 виділяють кілька підгруп пацієнтів. Основними транслокаціями є t(11;14)(q13;q32), t(4;14)(p16;q32), t(14;16)(q32;q23) та t(14;20)(q32;q12).
Декілька досліджень підтвердили, що пацієнти з ММ із транслокаціями t(4;14), t(14;16) і t(14;20) мають поганий прогноз, водночас t(11;14), як вважається, привносить менший ризик.
Делеція Del(13q) — поширена аномалія, яка спостерігається під час досліджень методом FISH, але є негативним прогностичним чинником, тільки якщо спостерігається під час метафазного цитогенетичного дослідження.
До частих хромосомних змін за ММ також належать аномалії хромосоми 117. У короткому плечі хромосоми найчастіше виникають делеції, у довгому — ампліфікації. Посилення/ампліфікація 1q21, а також делеція 1p підвищують ризик прогресування ММ, а частота виникнення ампліфікації вище в пацієнтів із рецидивами, ніж у пацієнтів із вперше діагностованим захворюванням.
Стратифікація пацієнтів на різні групи ризику залежно від хромосомних маркерів використовується деякими центрами в разі консультування з прогнозу захворювання, відбору пацієнтів і визначення послідовності застосування терапевтичних підходів. Метою застосування панелі FISH для прогностичної оцінки плазматичних клітин має бути виявлення наявності del 13, del 17p13, t(4;14), t(11;14), t(14;16), t(14:20), посилення/ампліфікації 1q21 і делеції 1p. Корисність цієї інформації полягає у визначенні біологічного підтипу, наданні прогностичних рекомендацій, а також у визначенні кандидатів для участі в клінічних дослідженнях.
Візуалізація
Протягом десятиліть оглядове рентгенологічне дослідження скелета була стандартом для оцінки враження кісток у будь-якої людини з підозрою на ММ. Проте цей метод має суттєві обмеження, пов’язані з нижчою чутливістю порівняно із сучасними методами візуалізації.
Було показано, що комп’ютерна томографія (КТ) окремо або в поєднанні з позитронно-емісійною томографією (ПЕТ) із фтордезоксиглюкозою (ФДГ) за чутливістю виявлення остеолітичних уражень достовірно переважає вказаний вище метод у пацієнтів із моноклональними плазмоклітинними захворюваннями. У багатоцентровому аналізі, виконаному IMWG, традиційне оглядове рентгенологічне дослідження скелета порівнювали з КТ усього тіла у 212 пацієнтів із моноклональними плазмоклітинними захворюваннями.
Результати КТ усього тіла були позитивними у 25,5 % пацієнтів із негативними результатами оглядового рентгенологічного дослідження скелета. Чутливість оглядового рентгенологічного дослідження скелета та низькодозової КТ усього тіла для довгих кісток достовірно не відрізняється, різниця переважно полягає у виявленні аномалій у хребті та тазі.
Крім того, дослідження показали, що КТ всього тіла з низькою дозою опромінення перевищує ефективність оглядової рентгенографії скелета в ділянках, які важко візуалізувати під час оглядового рентгенологічного обстеження скелета, як-от кістки черепа та ребра. Також було показано, що ПЕТ/КТ із ФДГ виявляє більше уражень, ніж звичайна рентгенографія, і виявляє ураження в пацієнтів із негативними результатами оглядового рентгенологічного дослідження скелета.
Слід відзначити, якщо замість низькодозової КТ усього тіла вибирається ПЕТ/КТ, якість візуалізації КТ-компоненту ПЕТ/КТ має бути еквівалентною низькодозовій КТ всього тіла. Зазвичай КТ компонент використовується тільки для корекції ослаблення, що може бути недостатнім для оцінки враження кісток внаслідок ММ і стабільності хребта. ПЕТ/КТ всього тіла корисна для виявлення екстрамедулярного враження поза межами хребта.
Для початкової діагностики пацієнтів із підозрою на ММ рекомендовано або низькодозову КТ всього тіла, або ПЕТ/КТ із ФДГ.
Зазначено, що оглядове рентгенологічне дослідження скелета, включно з довгими кістками, прийнятне у випадках, коли візуалізаційне дослідження із застосуванням сучасних методів недоступне (наприклад, в умовах обмежених ресурсів). КТ із контрастними речовинами не є необхідною для виявлення мієломного враження кісток і, за можливості, її слід уникати в пацієнтів із мієломою.
МРТ корисна для диференціації «тліючої» мієломи від ММ. Оскільки тяжкість захворювання в пацієнтів із «тліючою» мієломою нижча, ніж у пацієнтів із ММ, необхідно використовувати методи візуалізації з високою чутливістю, а МРТ є чутливим методом виявлення інфільтрації кісткового мозку мієломою.
Якщо результати низькодозової КТ всього тіла або ПЕТ/КТ із ФДГ негативні, слід розглянути можливість проведення МРТ всього тіла без контрастування, щоб відрізнити «тліючу» мієлому від ММ.
Для підтвердження наявності плазмоцитоми може також знадобитися біопсія тканин. Аналіз проліферації плазматичних клітин може бути корисним для визначення популяції клітин мієломи, що проліферують. Окрім того, за підозри на амілоїдоз діагноз встановлюється відповідно до рекомендацій, викладених у NCCN Guidelines щодо системного амілоїдозу легких ланцюгів. За підозри на клінічні симптоми підвищеної в’язкості слід оцінювати в’язкість сироватки, особливо в пацієнтів із високим рівнем М-білка. Якщо пацієнт розглядається як кандидат на алогенну трансплантацію, необхідно визначити його статус за системою людських лейкоцитарних антигенів (HLA-системою). Матричний аналіз однонуклеотидних поліморфізмів (single nucleotide polymorphism, SNP) та/або секвенування нового покоління (next generation sequencing, NGS) на відповідних панелях для матеріалу кісткового мозку допомагають провести більш детальну оцінку генетики ММ, що дає змогу додатково класифікувати ризик шляхом виявлення додаткових аномалій, які можуть мати прогностичне та/або терапевтичне значення.
Також рекомендовано ідентифікацію вихідного клону або зберігання зразка аспірату кісткового мозку для ідентифікації клонів для майбутнього тестування мінімальної залишкової хвороби (minimal residual disease, MRD) методом NGS, якщо це необхідно, а також оцінку плазматичних клітин, які циркулюють у периферичній крові, за клінічними показаннями.
Основною проблемою при лікуванні плазматичних новоутворень є відокремлення стабільної безсимптомної групи пацієнтів, які не потребують негайного лікування, від пацієнтів із прогресуючою симптоматичною мієломою, яких, можливо, потрібно лікувати негайно. Моноклональна гаммапатія невизначеного значення або тліюча мієлома повинні відрізняти від прогресуючої мієломи.
Безсимптомні плазмоцитинні новоутворення (тліюча множинна мієлома)
Безсимптомні пацієнти з множинною мієломою, які не мають літичних уражень кісток і мають нормальну функцію нирок, можуть спочатку безпечно спостерігатися поза контекстом клінічних випробувань. Наростаюча анемія є найбільш надійним показником прогресування. Наступні критерії представляють нове визначення тліючої мієломи.
- Сироватковий моноклональний білковий імуноглобулін (Ig) G або IgA не менше 30 г/л або моноклональний білок сечі не менше 500 мг на 24 години.
- Клональні плазматичні клітини кісткового мозку від 10% до 60% (>60% означає явну мієлому).
- Відсутність амілоїдозу або мієломних явищ, таких як:
- Гіперкальціємія більше ніж на 1 мг/дл вище норми.
- Креатинін більше 2 мг/дл або кліренс креатиніну менше 40 мл/хв.
- Анемія з гемоглобіном менше 10,0 г/дл.
- Ураження кісток (одне або більше) на рентгенографії скелета, комп’ютерній томографії (КТ) або позитронно-емісійній томографії (ПЕТ)-КТ.
- Відсоток клональних плазматичних клітин у кістковому мозку становить 60% або більше.
- Співвідношення залучений:незалучений легкий ланцюг без сироватки (FLC) становить 100 або більше.
- Більше одного вогнища ураження принаймні 5 мм на магнітно-резонансній томографії (МРТ) хребта.
Симптоматичні плазматичні новоутворення
Пацієнти з симптоматичним запущеним захворюванням потребують лікування.
Лікування найчастіше спрямоване на зменшення навантаження на пухлинні клітини та усунення будь-яких ускладнень захворювання, таких як ниркова недостатність, інфекція, підвищена в’язкість або гіперкальціємія, за допомогою відповідного медичного лікування. Міжнародна робоча група з мієломи (IMWG) опублікувала нові критерії для ідентифікації пацієнтів з активною мієломою, які потребують терапії. Ці критерії включають наступне:
- Амілоїдоз.
- Гіперкальціємія більше ніж на 1 мг/дл вище норми.
- Креатинін більше 2 мг/дл або кліренс креатиніну менше 40 мл/хв. Мієлома може викликати порушення функції нирок через гіперкальціємію, амілоїдоз або хворобу відкладення легких ланцюгів.
- Анемія з гемоглобіном менше 10,0 г/дл.
- Ураження кісток (одне або більше) на рентгенографії скелета, МРТ всього тіла або МРТ хребта та таза або ПЕТ-КТ.
- Відсоток клональних плазматичних клітин у кістковому мозку становить 60% або більше.
- Залучений: коефіцієнт FLC незалученої сироватки 100 або більше.
- Більш ніж одне вогнищеве ураження розміром принаймні 5 мм при огляді кісток скелета або, якщо результат негативний, МРТ всього тіла, або МРТ хребта та таза, або ПЕТ-КТ.
Прогноз
Множинна мієлома добре піддається лікуванню, але рідко виліковна. Середня виживаність в епоху прехіміотерапії становила близько 7 місяців. Після введення хіміотерапії прогноз значно покращився з середньою виживаністю від 24 до 30 місяців і 10-річною виживаністю 3%.
Покращення прогнозу відбулося завдяки впровадженню нових біологічних методів лікування та кращих варіантів порятунку, медіана виживаності зараз перевищує 60–90 місяців.
Множинна мієлома потенційно виліковна, якщо вона проявляється як одиночна плазмоцитома кістки або як екстрамедулярна плазмоцитома.
Не існує загальноприйнятої системи визначення стадії для моноклональної гаммопатії невизначеного значення, ізольованої плазмоцитоми кістки або екстрамедулярної плазмоцитоми. З плазматичних новоутворень система визначення стадії існує лише для множинної мієломи
Лікування
Сучасну терапію пацієнтів із симптоматичною мієломою можна розділити на такі категорії:
- Індукційна терапія.
- Консолідаційна терапія, яка менш застосовна для дуже літніх людей.
- Підтримуюча терапія.
Підтримуюча терапія, наприклад бісфосфонати.
Активна (симптомна) множинна мієлома Вперше діагностована ММ зазвичай чутлива до різних класів препаратів: імуномодуляторів (ІМ), інгібіторів протеасом (ІП) і моноклональних антитіл.
Первинна терапія активної (симптомної) множинної мієломи
Пацієнти з активною (симптомною) мієломою спочатку отримують первинну терапію, після чого застосовується високодозова хімієтерапія з аутологічною трансплантацією гемопоетичних клітин (ТГК) у пацієнтів, які відповідають критеріям трансплантації. Токсини стовбурових клітин, як-от нітрозосечовина, або алкілувальні засоби ставлять під загрозу резерв стовбурових клітин. Схем лікування з цими препаратами (зокрема, мелфаланом) слід уникати в пацієнтів, які є потенційними кандидатами на ТГК, доки в них не будуть зібрані стовбурові клітини.
Одним із перших кроків в оцінці пацієнтів із вперше діагностованою ММ є визначення того, чи є вони кандидатами на отримання високодозової терапії та трансплантації, залежно від віку та супутніх захворювань. Проте слід зазначити, що літній вік і порушення функції нирок не є абсолютними протипоказаннями до трансплантації. Тому важливо скерувати пацієнта до центру ТГК для оцінки того, чи відповідає він критеріям проведення ТГК.
Пацієнти молодше 65 років зазвичай вважаються молодшими та здоровими, тоді як пацієнти старше 75 років зазвичай не підлягають трансплантації. Супутні захворювання та працездатність є важливими визначальними факторами в будь-якому віці, особливо у віці від 65 до 75 років, які допомагають прийняти рішення про право на трансплантацію. Існують номограми для літніх пацієнтів, щоб визначити очікувану тривалість життя незалежно від діагнозу мієломи. Вік, дисфункція органів і ризик серцево-судинних і тромботичних ускладнень впливають на вибір індукційної терапії та розгляд консолідаційної терапії, такої як консолідація аутологічної трансплантації стовбурових клітин (SCT). Більшість пацієнтів також отримують ліки з бісфосфонатами або інгібіторами RANKL для запобігання ускладнень, пов’язаних із скелетом.
Для пацієнтів < 70 років у хорошому клінічному стані, індукція з подальшим проведенням терапія високими дозами АСТ є стандартним методом лікування.
Пріоритетні схеми первинної терапії для нещодавно виявлених кандидатів на трансплантацію включають бортезоміб/леналідомід/дексаметазон і бортезоміб/ циклофосфамід/дексаметазон.
Інші рекомендовані схеми первинної терапії для нещодавно виявлених кандидатів на трансплантацію Карфілзоміб/леналідомід/дексаметазон. Карфілзоміб — це інгібітор протеасом (ІП) другого покоління, який високоселективно та необоротно зв’язується з протеасомою. Він вводиться внутрішньовенно.
Через 2–4 місяців терапії пацієнтам, які відповідають на терапію, зазвичай проводять аутологічну консолідацію SCT. Після одужання після аутологічної SCT проводять підтримуючу терапію до моменту рецидиву. При рецидиві наступні терапії проводять послідовно, використовуючи раніше успішні препарати (якщо інтервал часу з моменту попереднього впливу становить >1 року) або новіші препарати, які раніше не пробувалися.
Пацієнти, які не є кандидатами на трансплантацію отримують індукційну хіміотерапію з триплетом (як описано для молодшого здорового пацієнта) плюс моноклональне антитіло до CD38, даратумумаб, або дублет і даратумумаб, які можуть краще переноситися. Терапія продовжується до досягнення максимальної відповіді, а потім застосовується підтримуюча терапія до рецидиву. При рецидиві наступні терапії застосовуються послідовно (як описано для молодшого здорового пацієнта).
Критерії оцінки відповіді на лікування IMWG
| Критерії відповіді на терапію | Критерії |
| Підтверджена повна відповідь | Нормальне співвідношення вільних легких ланцюгів і відсутність патологічного клона клітин у кістковому мозку за даними імуногістохімічного та імунофлюоресцентного досліджень |
| Повна відповідь | Негативна імунофіксація в сироватці та сечі, відсутність усіх м’якотканинних плазмоцитом; плазматичні клітини в кістковому мозку ≤5% |
| Дуже хороша часткова відповідь | М-градієнт у сечі та сироватці крові визначається лише методом імунофіксації або зниження М-градієнта в сироватці крові на ≥90% та М-градієнт у сечі <100 мг/добу |
| Часткова відповідь | Зниження М-градієнта в сироватці крові на ≥50% та зниження М-градієнта в добовій сечі на ≥90% або <200 мг/добу. Якщо М-градієнт у сироватці крові та сечі не визначається, критерієм оцінки може бути зменшення різниці рівнів патологічних і нормальних легких ланцюгів на ≥50%. Якщо вільні легкі ланцюги в сироватці крові визначити також неможливо, критерієм є зменшення плазматичних клітин на ≥50%, при умові, що вихідний вміст плазматичних клітин у кістковому мозку ≥30%. При наявності м’якотканинної плазмацитоми — зменшення її розмірів ≥50%. |
| Стабілізація захворювання | Не відповідає критеріям повної відповіді, дуже хорошої часткової відповіді, часткової відповіді або прогресування захворювання. |
Прямі ознаки загострення захворювання та/або порушення в органах-мішенях:
а) поява нових плазмоцитом м’яких тканин або нових кісткових уражень;
б) доведене збільшення в розмірах існуючих плазмоцитом або кісткових уражень;
в) доведене збільшення означає 50% (як мінімум 1,0 см) збільшення періодично вимірюваного діаметра вогнища ураження;
г) гіперкальціємія (>11,5 мг/100мл) (2,65 ммоль/л);
д) зниження рівня гемоглобіну ≥2 мг/100 мл.
Рецидив після ПВ (використовується лише, якщо кінцевою метою дослідження є визначення безподійного виживання) — будь-який один або декілька нижченаведених критеріїв: поява М-протеїну в сироватці крові та сечі при дослідженні методом імунофіксації чи електрофорезу; збільшення ≥5% плазматичних клітин у кістковому мозку; поява будь-яких інших ознак прогресії захворювання (це може бути нова плазмоцитома, літичні ураження кістки або гіперкальціємія).
Підтримуюча терапія
Пацієнти з мієломою, які відповідають на лікування, демонструють прогресуюче падіння білка М до досягнення плато; подальше лікування звичайними дозами не призводить до подальшого покращення. Це змусило дослідників поставити під сумнів, як довго слід продовжувати лікування. Жодне клінічне випробування не порівнювало напряму консолідаційний підхід із підтримуючим підходом, щоб оцінити, який є кращим для продовження ремісії та, зрештою, виживання. У більшості клінічних випробувань використовується один або обидва. Підтримуючі випробування з глюкокортикостероїдами та з інтерфероном показали дуже незначне покращення тривалості ремісії та виживання, але з токсичністю, яка переважала переваги. Ефективність і переносимість талідоміду, леналідоміду, бортезомібу та іксазомібу в умовах індукції та рецидиву зробила ці препарати привабливими варіантами в підтримуючих дослідженнях.
Усі випробування та мета-аналіз підтримуючої терапії леналідомідом показали значне покращення ВБП, тоді як ЗВ покращилася в одному дослідженні та одному мета-аналізі, обидва після аутологічної ТСК. Усі ці дослідження показали збільшення мієлодисплазії або гострого лейкозу з 3% до 7% для леналідоміду, що відповідає іншим дослідженням леналідоміду. Цей підвищений ризик здебільшого спостерігається у пацієнтів, які раніше застосовували алкілуючі агенти. Дози від 5 мг до 15 мг на день застосовувалися постійно або з 1 тижневою перервою щомісяця. Для пацієнтів, які не можуть отримувати підтримувальну терапію леналідомідом, розумною альтернативою є іксазоміб.
Після аутологічної ТСК пацієнтам пропонують підтримуючу терапію леналідомідом на основі постійної ВБП і випадкових переваг ОС, описаних раніше. Але короткострокова та довгострокова токсичність, а також фінансова токсичність можуть перешкодити реалізації. Пацієнти з високим ризиком, особливо з del(17p) або t(14;16), можуть потребувати підтримки бортезомібом (з або без леналідоміду), але цей підхід не ґрунтується на доказах, і необхідні підтверджуючі клінічні дослідження .
Лікування та профілактика мієломної хвороби кісток
Мієломна хвороба кісток є наслідком підвищеної активності остеокластів, і агенти, які інгібують остеокласти, є важливим компонентом терапії мієломи. Бісфосфонати памідронат і золедронат використовуються найчастіше шляхом внутрішньовенної інфузії, але інгібітор моноклональних антитіл RANKL деносумаб, який вводиться підшкірно, також ефективний, особливо коли дисфункція нирок не дозволяє використовувати бісфосфонати.
Бісфосфонати зазвичай вводять внутрішньовенно щомісяця протягом 2 років, а потім продовжують за тим самим або за скороченим графіком (тобто один раз кожні 3–4 місяці), якщо є докази активної мієломної хвороби кісток.
Деносумаб значно дорожчий, ніж бісфосфонати, які доступні в загальній формі.
На відміну від бісфосфонатів, оборотний механізм дії деносумабу може призвести до повторних переломів, якщо його припинити, хоча це теоретичне занепокоєння щодо пацієнтів з мієломою може бути пом’якшене безперервною підтримуючою терапією.
Рекомбінантні людський еритропоетин і дарбепоетин альфа можуть використовуватись для лікування мієлом-асоційованої анемії (рівень гемоглобіну <10 г/дл), коли інші причини анемії були виключено. Метою є підтримка гемоглобіну близько 12 г/ dl (нижче 14 г/дл, щоб уникнути тромбоемболічних ускладнень і гіпертонічної хвороби).
Лікування гранулоцитарним колонієстимулюючим фактором (G-CSF) може знадобитися для лікування важкої гранулоцитопенії, викликаної хіміотерапією. Інфекційні епізоди вимагають негайної терапії з антибіотиками широкого спектру дії. Профілактика інфекції залишається суперечливою, але може бути корисною протягом перших 2-3 місяців після початку терапії, особливо у пацієнтів, які отримують леналідомід або помалідомід або у пацієнтів з високим ризиком інфікування (попередній серйозні інфекції або нейтропенія). Рекомендуються щеплення від грипу та пневмокока.
Ацикловір або валацикловір для профілактики вірусу герпесу рекомендований для пацієнтів, які отримують терапію на основі інгібіторів протеасом. Імуноглобулінна профілактика рутинно не рекомендується.Мова перекладу: Українська
Венозна тромбоемболія. У пацієнтів з ММ спостерігається підвищений ризик тромбозу з початковим ризиком 3%–4% венозних тромботичних подій, і цей ризик значно підвищується в умовах терапії із застосуванням специфічних препаратів. Високі дози дексаметазону, цитотоксична хіміотерапія, така як доксорубіцин і IMiDs (талідомід і lenalidomide) істотно підвищують цей ризик. Інші фактори, такі як зниження рухливості внаслідок неврологічних ускладнень або болю в кістках, супутні переломи, одночасне застосування еритропоез-стимулюючих агентів та попередній особистий або сімейний анамнез тромботичних подій підвищують ризик тромбоемболічних подій. Поточні рекомендації для пацієнтів з ММ, які повинні розпочати IMiD-терапію, полягають у тому, щоб застосовувати аспірин (100 мг) при відсутності факторів ризику тромбозу і застосовувати повні дози антикоагулянтів для осіб з підвищеним ризиком (низькомолекулярні гепарини або варфарин).
Променева терапія при ураженнях кісток
ММ — високорадіочутлива пухлина, однак променеву терапію як самостійний метод використовують дуже рідко. Дози, що використовують, коливаються у широких межах — 35–50 Гр за 4–5 тиж. У більшості випадків променеву терапію призначають на зони можливих патологічних переломів у дозі від 15–30 Гр., як метод профілактики їх виникнення або на зони існуючих переломів для їх консолідації.
Локальна променева терапія також ефективна для зменшення вираженості болю в кістках. Згідно з результатами досліджень при застосуванні променевої терапії зменшення вираженості болю відмічалось у 91–97% пацієнтів, в тому числі повне зникнення больового синдрому — у 21–26% пацієнтів, які отримали фракціоновану променеву терапію
Літичні ураження хребта зазвичай вимагають опромінення, якщо є будь-який із наведених нижче фактів:
- Вони пов’язані з екстрамедулярною (параспінальною) плазмоцитомою.
- Виникла хвороблива деструкція тіла хребця.
- КТ або МРТ показують стиснення спинного мозку.
Біль у спині, викликана остеопорозом і невеликими компресійними переломами хребців, найкраще піддається хіміотерапії.
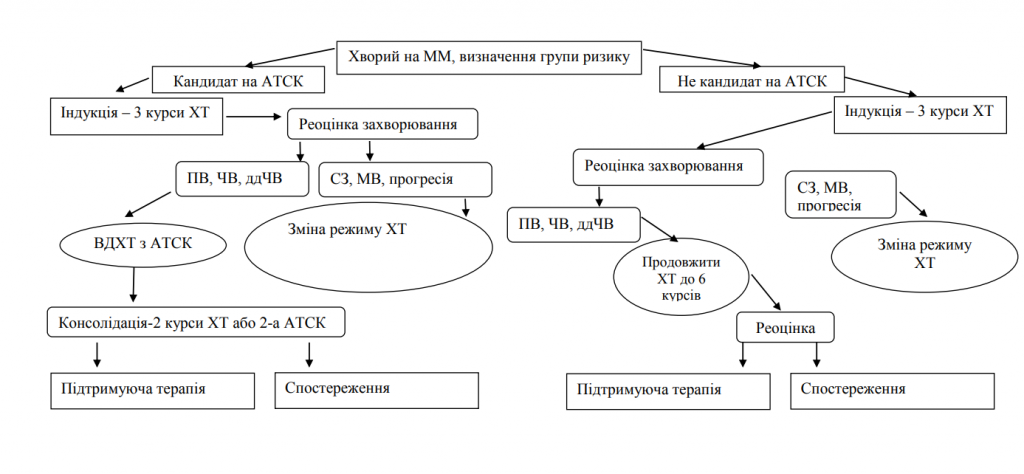
Алгоритм лікування хворих на множинну мієлому
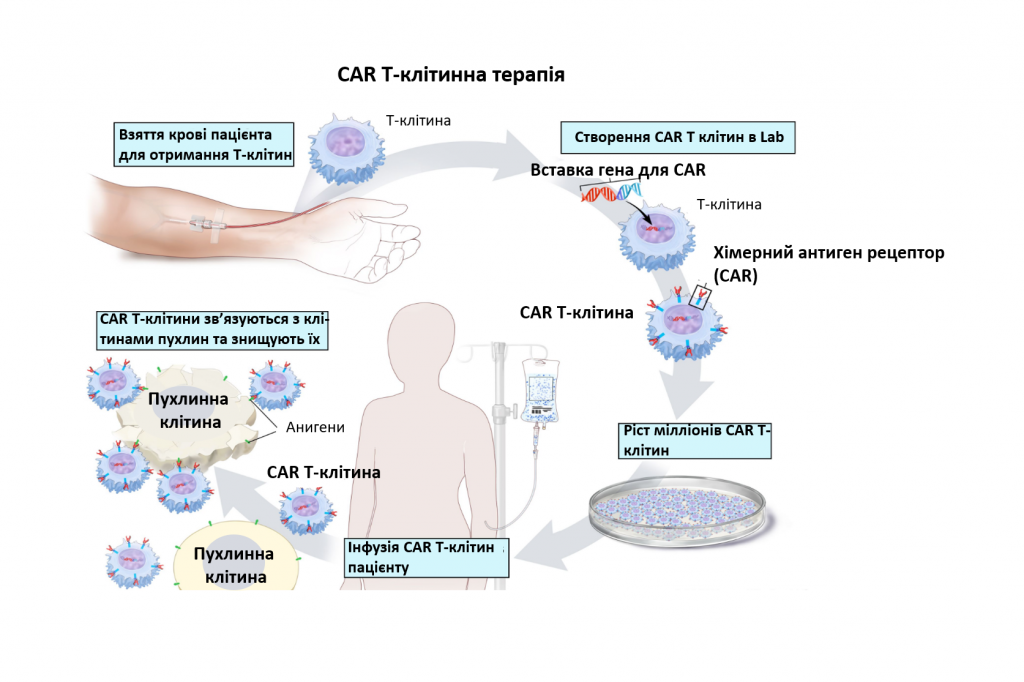
Т-клітинна терапія CAR
Т-клітинна терапія CAR: це лікування змінює Т-клітини пацієнта (тип клітин імунної системи), щоб вони атакували певні білки на поверхні ракових клітин. У пацієнта беруть Т-клітини і в лабораторії на їх поверхню додають спеціальні рецептори. Змінені клітини називають Т-клітинами рецептора химерного антигену (CAR). Т-клітини CAR вирощують у лабораторії та вводять пацієнту шляхом інфузії. Т-клітини CAR розмножуються в крові пацієнта і атакують ракові клітини.
Т-клітинна терапія CAR вивчається для лікування множинної мієломи яка рецидивувала (повернулася).
Зареєструйтеся на нашому сайті прямо зараз, щоб мати доступ до більшої кількості навчальних матеріалів!
Підписатися на наші сторінки:
Джерела:
- Bradford A. Curt, Charles Kuntz, IV, Ferhan A. Asghar Differential Diagnosis and Initial Management Spine Pathology
- Frederick R. Singer Paget’s Disease of Bone
- https://www.healthdirect.gov.au/pagets-disease-of-bone
- NCCN Guidelines
- Oi Lin Lee, Noemi Horvath, Cindy Lee, Doug Joshua, Joy Ho, Jeff Szer, Hang Quach, Andrew Spencer, Simon Harrison, Peter Mollee, Andrew W. Roberts, Dipti Talaulikar, Ross Brown, Bradley Augustson, Silvia Ling, Wilfrid Jaksic, John Gibson, Anna Kalff, Anna Johnston, Akash Kalro, Chris Ward, H. Miles Prince, Andrew Zannettino Bisphosphonate guidelines for treatment and prevention of myeloma bone disease
- P. Moreau, J. San Miguel, P. Sonneveld, M. Attal at al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up DOI:https://doi.org/10.1093/annonc/mdx096
- National Cancer Institute Plasma Cell Neoplasms (Including Multiple Myeloma) Treatment (PDQ®)
- Multiple myeloma
- Dimopoulos, Meletios A.; Moreau, Philippe; Terpos, Evangelos; Mateos, María-Victoria; Zweegman, Sonja; Cook, Gordon; Delforge, Michel; Hájek, Roman; Schjesvold, Fredrik; Cavo, Michele; Goldschmidt, Hartmut; Facon, Thierry; Einsele, Hermann; Boccadoro, Mario; San-Miguel, Jesús; Sonneveld, Pieter; Mey, Ulrich Multiple Myeloma: EHA-ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up DOI: 10.1097/HS9.0000000000000528
- Aula Ramo, Cortney Mckay, Vrushali Dabak Cardiac Considerations With Carfilzomib in a Patient Diagnosed With Multiple Myeloma
- Laura Rosiñol, Albert Oriol, M Jesús Blanchard, Luis Palomera, Maria-Victoria Mateos, Javier De La Rubia, Miguel T Hernandez, Joaquín Díaz-Mediavilla, Jose Mariano Hernandez, Raquel Jiménez, Cristina Motllo, Juan José Lahuerta, Jesus F. San Miguel, Joan Blade Tumor and Renal Response in Patients with Newly Diagnosed Multiple Mieloma and Renal Failure Treated with Bortezomib and Dexamethasone: Results of a Prospective Phase II Trial from Pethema/GEM https://doi.org/10.1182/blood.V124.21.4776.4776
- https://empendium.com/ua/chapter/B27.II.15.15

Гостра мієлоїдна лейкемія
Гостра мієлоїдна лейкемія (ГМЛ) включає гетерогенну групу агресивних ракових клітин крові, які виникають внаслідок клональної…

Консультування пацієнта при підозрі на гостру мієлоїдну лейкемію – рекомендації ОСКІ
Початок консультації Вимийте руки та одягніть ЗІЗ, якщо це необхідно Представтесь пацієнту, вказавши своє ім’я…

Консультування пацієнта з хронічною мієлоїдною лейкемією – рекомендації OSCE
Автори: Юлія Малишева, Дмитро Гамов, Наталія Лопіна Початок консультації Вимийте руки та одягніть ЗІЗ,…
Хронічна мієлоїдна лейкемія
Хронічна мієлоїдна лейкемія – це мієлопроліферативне новоутворення, що характеризується нерегульованою продукцією та неконтрольованою проліферацією зрілих…

Обстеження лімфоретикулярної системи / рекомендації ОСКІ
Оцінку лімфатичної системи слід проводити як частину комплексної оцінки, як під час звичайного об’єктивного огляду,…
